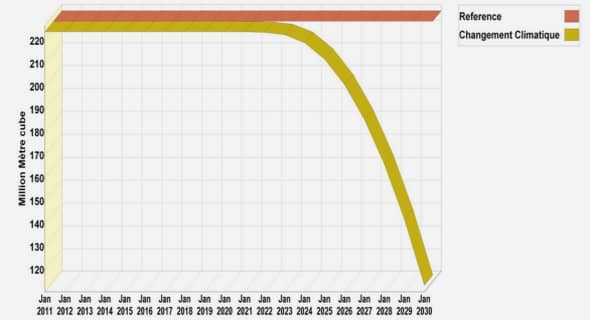
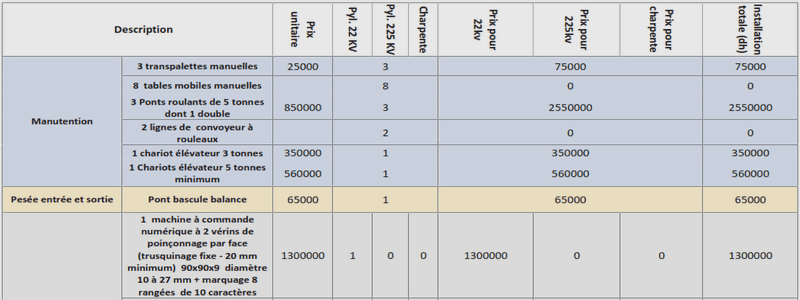
Au départ de la 3,6-dichloropyridazine
Synthèse de dihydropyrrolopyridazines
Préparation du précurseur xanthate
La préparation du précurseur xanthate (cf. schéma IX-3) a débuté avec la substitution en tube scéllé de la 3,6-dichloropyridazine 1 par la tert-butylamine pour donner l’amine IX-01 avec un rendement maximum très faible de 24%, principalement à cause d’une nette dégradation du milieu réactionnel. Des conditions plus douces comme un chauffage avec ou sans reflux, dans le N,N-diméthylacétamide (cf. les travaux sur les dérivés de la pentachloropyridine IV-28 et IV-29) pourraient être une meilleure alternative, bien que nous ne les ayons pas testées. L’étape suivante d’acétylation par du chlorure de chloroacétyle dans le toluène au reflux permet d’obtenir IX-02 avec un rendement de 90%. Le chlore est ensuite déplacé par du sel d’O-éthyl xanthogénate de potassium pour donner avec un rendement de 99% le précurseur attendu IX-03. Un point qu’il convient de noter est le manque de reproductibilité des rendements de cette synthèse, c’est pourquoi nous avons donné des fourchettes de rendement, et non uniquement le rendement maximum obtenu.
Essais de réactions radicalaires de cyclisation et d’addition
L’étape finale de cyclisation radicalaire a été réalisée dans le chlorobenzène au reflux avec comme initiateur le DLP (cf. schéma IX-4). Cependant, à notre grande déception, aucune trace du produit IX-04 n’a pu être détectée. Parmi les nombreux sous-produits présents dans le milieu réactionnel, seul le composé de réduction du xanthate a pu être identifié. De la même façon l’addition intermoléculaire sur l’acétate d’allyle n’a pas permis d’obtenir le produit attendu IX-05, ni aucun autre produit identifiable. Une explication possible à ce manque de résultats (cf. schéma IV-5) vient des possibilités d’évolution du radical 4 (en équilibre avec son rotamère 5), qui peut au choix : • Cycliser de façon ipso sur le carbone 4 pour conduire à l’intermédiaire 6, qui après oxydation et ré-aromatisation, conduit au produit attendu IX-04. Cette voie réactionnelle est cinétiquement limitée par l’étape d’oxydation du radical 6 par le DLP. • Ou bien cycliser de façon ipso sur l’azote en position 2 pour conduire à l’intermédiaire 8, en équilibre avec les formes 9 et 10. Cependant aucun de ces trois intermédiaires ne peut conduire, via un enchaînement oxydation/ré-aromatisation, à un produit stable. Il s’agit d’un radical persistant, dont le seul devenir possible est la dimérisation ou bien la dégradation. Le problème est que l’étape de cyclisation sur l’azote menant de 5 à 8 n’est probablement pas réversible. En effet la liaison carbone-azote dans 8 est relativement haute en énergie, beaucoup plus que la liaison aromatique azote-azote de la pyridazine cassée lors de la cyclisation, et dans ces conditions le retour vers 5 n’est pas possible. De plus, si ce retour était bien possible, alors l’ensemble de l’équilibre serait drainé petit à petit avec l’avancement de la réaction d’oxydation conduisant de 6 à 7 pour finalement mener au produit IX-04. La liaison azote-azote agit donc comme un piège à radical, pour preuve, la réaction d’addition intermoléculaire sur l’acétate d’allyle conduit rapidement à un milieu réactionnel dégradé comme pour l’essai de cyclisation. Le radical n’a pas le temps d’effectuer l’addition intermoléculaire qu’il est piégé par la liaison azote-azote du cycle. Au vu de ces résultats nous avons décidé de ne pas poursuivre nos travaux au départ de la 3,6-dichloropyridazine, en particulier concernant la synthèse de dihydropyrrolopyridazines via la substitution d’allylamine en position 3.Ces travaux sur les trichloropyrimidines ont été repris par la suite par le docteur Petit lors de sa thèse. Tous les résultats issus de son travail et présentés dans ce chapitre seront clairement indiqués comme tels. Les travaux ont été menés au départ de la 2,4,6-trichloropyrimidine 1 (cf schéma X-1). Ce précurseur permet de reproduire le motif de la 2,6-dichloropyridine précédemment rencontré, de façon symétrique qui plus est, et donc d’évaluer la possibilité de cyclisation radicalaire sur un azote aromatique. Deux voies d’études sont possibles : • Soit à partir d’allylamine substituée en position 2 ou 4 pour conduire aux noyaux dihydroimidazopyrimidinone 2 et 4, et dihydropyrrolopyrimidine 3. • Soit à partir de tert-butylamine ou de méthylamine substitutée en position 2 ou 4 pour conduire aux de noyaux dihydropyrrolopyrimidinone 6 et imidazopyrimidinedione 5 et 7 Il est important de remarquer que pour les molécules issues d’une réaction de cyclisation sur azote (composés 2, 4, 5 et 7), il est possible d’obtenir a priori les deux isomères a et b, comme nous le verrons ci-après. Nous présenterons d’abord les résultats issus de l’utilisation d’allylamine en position 2 ou 4 (paragraphe 1), puis ceux issus de l’utilisation de tert-butylamine et de méthylamine en position 2 ou 4 (paragraphe 2).
Synthèse de dihydroimidazopyrimidinones & dihydropyrrolopyrimidines
Préparation des précurseurs oléfines
La préparation des précurseurs oléfines (cf. schéma X-2) commence avec la subsitution de la trichloropyrimidine par l’allylamine dans l’éthanol selon le protocole décrit par Botta et al.290 . Deux régioisomères sont obtenus, le produit minoritaire X-01 en position 2, avec un rendement de 28%, et le produit majoritaire X-02 en position 4, avec un rendement de 56%, pour un rendement global de 84%. A noter que ces conditions donnent un rendement limité à cause de la polysubstitution du composé de départ. Il serait sans doute profitable d’appliquer les même conditions que pour les pentachloropyridines (i.e. réaction dans le N,N-diméthylacétamide, sans être à reflux, avec entre un et deux équivalents d’allylamine). Les deux oléfines X-01 et X-02 sont ensuite protégées, soit par un groupe acétyle, soit par un carbamate de tert-butyle. Dans le cas de la protection sous forme de Boc, les deux composés X-03 et X-06 sont obtenus avec de bons rendements, respectivement 90% et 77%. Chapitre X Au départ de la 2,4,6-trichloropyrimidine 199 Cependant l’acétylation par le chlorure d’acétyle s’est avérée problématique (cf. tableau X-1). L’utilisation de conditions relativement douces à base de 4-diméthylaminopyridine catalytique dans le dichlorométhane au reflux (cf. entrées 1 et 2) se sont avérées insuffisantes pour convertir efficacement les substrats (0% à partir de X-01 et 31% à partir de X-02, avec récupération des composés de départ correspondants). Cela peut s’expliquer dans les deux cas par le fort appauvrissement électronique du cycle pyrimidine à cause des deux chlores restants. C’est particulièrement vrai pour X-01, ce qui explique l’absence totale de produit.
Addition/cyclisation à partir de la position 2 L’étape suivante est celle d’addition radicalaire intermoléculaire sur les précurseurs oléfines X-03 et X-04 (cf. schéma X-3 et tableau X-2). Un premier essai d’addition du xanthate Xa1 sur l’oléfine X-03 a permis d’obtenir le produit d’addition X-07 avec un rendement modéré de 59%, qui peut s’expliquer par plusieurs facteurs : • Le produit d’addition déprotégé est présent en quantité non négligeable (rendement isolé >10%). • Le produit de cyclisation X-10 est lui aussi obtenu, mais en quantité plus faible (>5% en RMN du brut). • La polarité très faible du composé de départ X-03 fait que le polarité du produit d’addition est entièrement dictée par celle du xanthate utilisé Xa1, rendant leur séparation par chromatographie très difficile s’il reste du réactif xanthate (ce qui est le cas ici, puisque 2 équivalents sont utilisés).