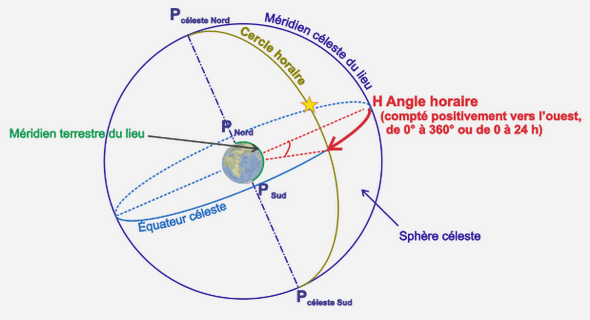
Généralités sur l’organocatalyse
Définition de l’organocatalyse
Le terme organocatalyse désigne la catalyse des réactions chimiques où le catalyseur utilisé pour accélérer la réaction est une petite molécule organique, dépourvue d’éléments inorganiques tels que des métaux, un tel catalyseur est appelé organocatalyseur. Bien que l’utilisation de molécules organiques en tant que catalyseurs soit connue depuis les débuts de la chimie, c’est uniquement au cours de ces dernières décennies que l’organocatalyse s’est démarquée et est devenue un concept à part entière [2]. Elle fait partie des grandes familles de catalyse homogène, aux côtés de la catalyse par les métaux ainsi que la catalyse par les enzymes .
Evolution de l’organocatalyse
Le terme organocatalyse a été inventé en 1935 par le chimiste allemand Wolfgang Langenbeck [4]. Les organocatalyseurs sont généralement constitués d’atomes comme le carbone, l’hydrogène, le soufre, le phosphore, l’azote, l’oxygène (C, H, S, P, N, O, …). La plupart du temps, l’organocatalyseur est une molécule chirale énantiopure et permet d’obtenir un produit chiral énantioenrichi: on parle alors d’organocatalyse énantiosélective. En outre, on parlera d’organocatalyse asymétrique si l’organocatalyseur est chiral et permet de contrôler la configuration des centres stéréogènes créés lors de la réaction. L’organocatalyse s’est développée très rapidement depuis 2000 compte tenu du nombre de publications noté dans ce domaine par an. Il passe de moins de 100 publications avant 2003 à plus de 1000 après 2010 comme le montre le graphe ci-dessous (Schéma 3).
Types d’organocatalyseurs
La majorité des catalyseurs organiques qui viennent d’être découverts sont à base d’amines chirales (acides aminés, peptides, alcaloïdes comme la quinine de cinchona, les imidazolidinones chiraux) [5]. Les organocatalyseurs sont souvent regroupés par caractère acido-basique. On distingue ainsi les bases et les acides de Lewis, les bases et les acides de Bronsted. Une autre classification est possible selon les différents modes d’action: la catalyse par l’intermédiaire d’énamines, d’ions iminiums, de carbènes, l’activation par liaisons hydrogène, la catalyse SOMO (plus haute orbitale moléculaire occupée) et la catalyse par l’intermédiaire du contre-ion . principaux modes d’activation sont détaillés ci-après.
Catalyse par intermédiaire d’énamines
Ce type de catalyse est mis en jeu lors de réactions de fonctionnalisation en α de composés carbonylés. Dans le mécanisme proposé (schéma 4a), le catalyseur A, une amine primaire ou secondaire, réagit avec le composé carbonylé pour former un ion iminium B, en équilibre tautomérique avec l’énamine C. Après réaction avec un électrophile (E+), un nouvel ion iminium D est formé. L’hydratation de ce dernier permet de récupérer le produit et de régénérer le catalyseur. La force motrice de cette réaction est la formation d’un intermédiaire énamine fortement nucléophile. Diverses transformations chimiques utilisent l’énamine en tant que nucléophile, comme par exemple les réactions de Michael, de Mannich ou les aldolisations. Elle peut également jouer le rôle de diénophile dans les réactions de cycloaddition de type Diels- Alder. Ainsi, de nombreux catalyseurs chiraux ont été développés afin d’étudier et d’améliorer l’efficacité et la sélectivité de ces transformations (schéma 4b) [7].de la LUMO) permettant une réaction de déprotonation afin de conduire à l’énamine correspondante. Cette entité nucléophile peut alors réagir avec des entités électrophiles permettant ainsi de réaliser des additions ou des substitutions nucléophiles. L’hydratation de l’iminium résultant permettra de régénérer le catalyseur en libérant le nouveau dérivé carbonylé. Le mécanisme de formation d’énamine est universel pour les amines secondaires et primaires et par conséquent le terme de catalyse énamine a été attribué à ce type d’activation [9]. Il intervient principalement dans les réactions d’aldolisation et de Mannich asymétriques (schéma 6) .
Catalyse par liaison hydrogène
La liaison hydrogène est une interaction attractive entre le doublet libre d’un atome fortement électronégatif (N, S, O…) et un atome d’hydrogène lié de façon covalente à un atome donneur électrons (également fortement électronégatif). Par le passé, les liaisons hydrogènes étaient considérées ne pas être suffisamment activantes ou directionnelles pour être utilisées en synthèse asymétrique. Le concept de la catalyse par liaison hydrogène fut découvert suite à l’observation d’une augmentation de la cinétique des réactions de Diels – Alder après ajout d’additifs protiques tels que des acides carboxyliques ou des phénols [11]. Cependant, son potentiel ne fut réalisé qu’au début des années 80 alors que Hiemstra décrivit les premièresadditions conjuguées catalysées par un alcaloïde bifonctionnel, la cinchonidine [12]. Ces travaux furent suivis par ceux de Jacobsen et de Corey qui élaborèrent les premiers catalyseurs interagissant par liaisons hydrogène à partir de l’hydrocyanation respective d’aldéhydes et d’imines [13]. Depuis, divers catalyseurs furent élaborés et exploités en synthèse asymétrique. Parmi les plus représentés, nous pouvons citer les urées et surtout thiourée, les guanidines et amidines, les diols et biphénols, les acides phosphoriques chiraux, les alcaloïdes dérivés de quinquina, les oligopeptides et plus récemment les squaramides.