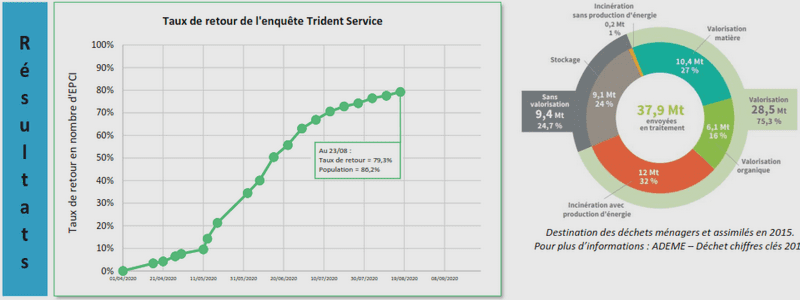
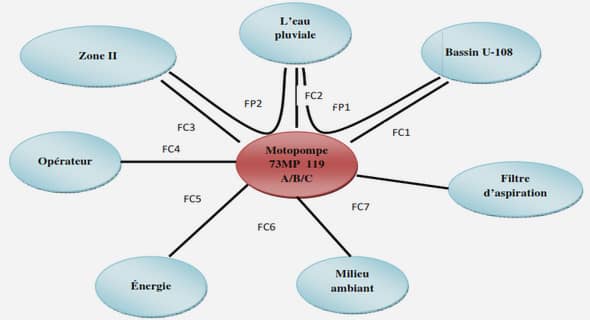
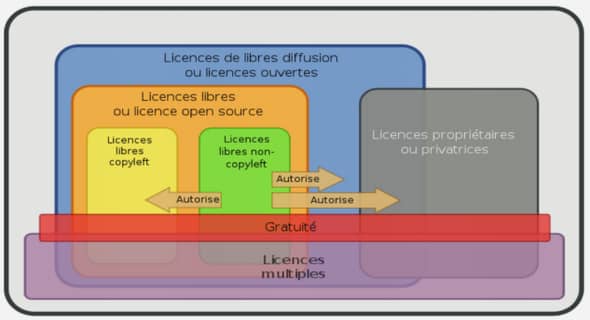
METHODES ELECTROCHIMIQUES POUR L’ANALYSE IN SITU DE COMPOSES BIOACTIFS EN MILIEU OCEANIQUE
Fabrication des électrodes
La réalisation des électrodes indicatrices nécessite un protocole précis de préparation. Nous avons testé deux types de fabrication : par chronoampérométrie et par trempage. Le premier nécessite une étape de voltammétrie cyclique pour le nettoyage du fil d’argent puis pour leur détection par réduction. La seconde méthode est plus simple : le fil d’argent est trempé dans une solution de Na2S très concentrée pendant 12 heures. Le nettoyage du fil d’argent a pour but d’éliminer tout autre élément susceptible de contamination. Il est réalisé par voltammétrie cyclique à la suite de nombreux balayages de potentiel (jusqu’à obtention d’un signal stable) ; le fil d’argent est placé dans de l’acide sulfurique 5N. IV. Mesures électrochimiques des sulfures dans les écosystèmes profonds océaniques Une fois le fil propre, on utilise à nouveau la voltammétrie cyclique pour le repérage du potentiel d’oxydation et de réduction des ions sulfure (Figure IV.1). Le fil d’argent est placé dans une solution de sulfure (760 µmol/l) tandis que le potentiel appliqué à l’électrode de travail (Ag) par rapport à celui de l’électrode de référence (Ag/AgCl) est balayé de -0.4 à 0.9 V. Deux pics apparaissent : l’un correspondant à l’oxydation de l’argent, l’autre à la réduction des ions sulfure. Pour une concentration de 760 µmol/l ces pics se situent respectivement autour de -570 mV et – 780 mV. Figure IV.1 : Détection des pics d’oxydation et de réduction pour la préparation d’une l’électrode (C [H2S]t = 760 µmol/l). Deux méthodes de préparation des électrodes, par chronoampérométrie et par trempage de l’électrode et réaction spontanée des ions sulfure avec l’argent, ont été testées ainsi que plusieurs conditions de stockage. La comparaison des résultats obtenus en fonction de la préparation et du stockage doit permettre d’optimiser le protocole pour la réalisation et la conservation des électrodes. La réaction qui se produit à la surface de l’électrode lors de sa création est la suivante : Réaction IV.1 2Ag+S2- → Ag2S + 2 eQuatre électrodes ont été préparées par chronoampérométrie, numérotées 1, 2, 10, 11 et quatre électrodes par trempage, numérotées 12, 13, 14 et 15. -225 -150 -75 0 75 150 -1 -0.8 -0.6 -0.4 E (V vs Ag/AgCl) I (µA) Oxydation Réduction Ag2S + 2 e- → 2Ag + S2- 2Ag+S2-→ Ag2S + 2 e-
Etude de la réponse des électrodes à détection potentiométriques dans le temps
Chrono ampérométrie
Le fil d’argent à recouvrir de sulfure d’argent est placé dans l’eau de mer artificielle contenant environ 700 µmol/l de sulfure. Un potentiel constant de -575 mV, par rapport à l’électrode de référence Ag/AgCl, est imposé. La durée du dépôt est au moins égale à 500 secondes.
Trempage
Deux électrodes ont été réalisées par trempage : le fil d’argent est laissé dans une solution très concentrée d’ions sulfure (environ 10 mmol/l) pendant 12 heures. La réaction entre l’argent et les ions sulfure est spontanée et conduit à la formation de Ag2S. Deux hypothèses sont proposées pour cette réaction spontanée : le bilan des Réaction IV.2 et Réaction IV.3 et le bilan des Réaction IV.4 etRéaction IV.5.
Détection des ions sulfure
Les gammes étalons sont préparées selon la méthode des ajouts dosés. La mesure est réalisée en présence d’une électrode combinée de température et de pH, afin de déterminer les concentrations des différentes espèces de soufre -II et leur quantité dans la solution. Les mesures sont également faites avec une agitation pour homogénéiser la solution le plus rapidement possible. Un exemple d’étalonnage de l’électrode n° 1 est présenté ci –après. La Figure IV.2 montre le potentiel mesuré en fonction du temps lors d’ajouts successifs de solution mère de sulfure : chaque palier correspond à un ajout. -0.7 -0.6 -0.5 -0.4 -0.3 -0.2 0 300 600 900 1200 Temps (s) E (V) 4 µmol/L [H2S]t 13 µmol/L [H2S]t 40 µmol/L [H2S]t Figure IV.2 : Suivi du potentiel en fonction du temps lors d’un étalonnage réalisé par ajouts successifs de solution mère d’ions sulfure. On note une stabilisation du signal très rapide (<1 seconde) pour des concentrations supérieures à 20 µmol/l. La Figure IV.3 présente l’étalonnage correspondant pour des concentrations en S2- entre 0.03 et 12 nmol/L, correspondant à des concentrations en sulfure total entre 40 et 900 µmol/l (calculés grâce aux constantes d’acidité données dans le Chapitre I). Selon les électrodes et leur mode de stockage la gamme de concentrations de sulfure total mesuré peut varier entre 4 µmol/l et 13 mmol/l. La pente, de l’ordre de 32 mV/ décade, est proche de la valeur théorique. IV.2. Etude de la réponse des électrodes à détection potentiométriques dans le temps 125 y = -14.175Ln(x) – 599.57 R2 = 0.9998 -640 -620 -600 -580 -560 -540 0.0 0.1 1.0 10.0 100.0 conc S 2- nmol/L E (mV) Figure IV.3 : Etalonnage de l’électrode 1 pour des concentrations en S2- entre 0.03 et 12 nmol.l-1, soit une concentration en sulfure total entre 40 et 900 µmol/l. Au niveau des écosystèmes profonds, la concentration en ions sulfure fluctue rapidement, c’est pourquoi il est important que le temps de réponse des électrodes soit minimal quelle que soit la concentration du milieu. Sur la Figure IV.2 le signal n’est pas stable pour les faibles concentrations (<40 µmol/l) et présente une instabilité qui n’est pas retrouvée pour les fortes concentrations. Cette réponse dépend toutefois de l’électrode et de son mode de stockage, en général plus l’électrode est utilisée, plus sa gamme de linéarité est étendue et sa limite de détection basse. La Figure IV.4 ci-dessous montre la stabilité du signal sur dix minutes pour une concentration d’environ 600 µmol/l. Figure IV.4 : Stabilité signal sur 10 minutes. 090506essais_stabilité_elect1_10min -650,0 -625,0 -600,0 -575,0 -550,0 1225 1325 1425 1525 1625 1725 1825 temps (s) E(mV)
Mesures électrochimiques des sulfures dans les écosystèmes profonds océaniques
Le signal observé est parfaitement stable avec une moyenne et un écart-type respectivement de -634,38 mV et 0,15 mV, soit une précision (définie comme 2 écarts types) de 0,05%.
Reproductibilité
Les échantillons sont préparés dans le but d’effectuer un test de reproductibilité. Un étalonnage est effectué avant le dosage des échantillons. Les 10 échantillons, tous issus de la même solution, sont préparés dans différents béchers. Deux tests de reproductibilité ont ainsi été réalisés sur deux jours successifs avec la même électrode (n°1) (Figure IV.5). Réplicabilité électrode 1 500 550 600 650 700 750 0 1 2 3 4 5 6 7 8 9 10 N° de l’échantillon Concentration (µmol/L) le 11/05/06 le 16/05/06 Figure IV.5 : Tests de reproductibilité : suivi de la réponse de 10 échantillons de même concentration (autour de 690 µmol/l) à deux reprises. Les variations de concentration mesurées entre échantillons sont très faibles, les précisions (calculées à partir de 2 écarts types) sont inférieures à 3 % (Tableau IV.2). La méthode apparaît reproductible à plusieurs jours d’intervalle. Tableau IV.2 : Test de reproductibilité. Date Concentration moyenne (µmol/l) Ecart type (σ) Précision (2 σ) le 11/05/06 688.6 4.7 1.4 % le 16/05/06 694.8 10.,2 2.9 %
Etude de la réponse des électrodes à détection potentiométriques dans le temps
Etude de la stabilité dans le temps des électrodes
L’évolution de la concentration à partir de laquelle la réponse de l’électrode à la concentration de S2- devient linéaire a été suivie pour chacune des électrodes. La sensibilité des électrodes dans la gamme de linéarité a également été déterminée au cours du temps (Tableau IV.3). La Figure IV.6 illustre l’évolution des droites de calibration pour l’électrode n° 2 en fonction du temps, pendant la période du 4 mai 2006 au 9 août 2006 (soit 97 jours après sa fabrication). -660 -630 -600 -570 -540 0.01 0.10 1.00 10.00 100.00 conc S2- nmol/L E (mV) 04 05 06 07 07 06 13 07 06 20 07 06 25 07 06 09 08 06 Figure IV.6 : Evolution des droites de calibration de l’électrode n°2 en fonction du temps pendant 97 jours. Cette électrode a été préparée par chronoampérométrie et stockée à l’air. On note que les pentes des droites de calibration sont proches de 30 mV/ décade et donc peu différentes de la valeur théorique (29 mV/décade). Ce décalage pourrait s’expliquer par le fait que la température est mesurée mais pas totalement contrôlée (entre 20 et 25 °C), et /ou par le fait que lors de la formation du sulfure d’argent à la surface de l’électrode d’autres composés peuvent également être formés (polysulfures…).
LISTE DES PRINCIPAUX SIGLES ET ABREVIATIONS UTILISES |