Evaluation de la prophylaxie à faibles doses avec des concentrés de facteurs de coagulation à demi-vie prolongée dans l’hémophilie
Notions de base
L’hémophilie est une maladie hémoragique héréditaire à transmission recessive liée au sexe (le gène des facteurs est localisé sur le chromosome X) due à une anomalie moléculaire au niveau des gènes des facteurs VIII et IX. Elle est caractérisée par un déficit partiel ou total en facteur de la coagulation VIII (hémophilie A) ou IX (hémophilie B). Sa sévérité clinique est fonction de l’importance du déficit en ces facteurs faisant distinguer 3 formes cliniques : majeure (taux < 1%), modérée (taux entre 1et 5%) et mineure (taux entre 6 et 30%)
Epidémiologie
Hémophilie dans le monde
L’hémophilie est une maladie hémorragique constitutionnelle ubiquitaire avec une incidence d’un cas pour dix-mille naissances sans prédilection de race ou de région géographique. La population qui souffre d’hémophilie dans le monde est estimée à un demi-million de personnes, soit une prévalence de 1 cas pour 5000 naissances pour l’hémophilie A et 1 cas pour 25000 naissances pour l’hémophilie B (ratio de 4 pour 1 entre ces deux types). Seuls 191216 hémophiles sont connus d’après le rapport annuel de la FMH de 2018 (40, 77, 85). I.2.2. Hémophilie en Afrique La prévalence de l’hémophilie en Afrique est très mal connue variant entre 0,8 et 1,64 (64) ; cependant, en 2017, seuls 2,83% des cas diagnostiqués dans le monde provenaient d’Afrique (85). Les données publiées concernent surtout l’Afrique du Nord et l’Afrique anglophone (78). Des données récentes montrent une nette amélioration de la prise en charge de l’hémophilie en Afrique sub-saharienne
Hémophilie au Sénégal
La prévalence de l’hémophilie au Sénégal est estimée à 2,3 cas pour 100000 naissances de sexe masculin avec un âge médian au diagnostic de 4,2 ans. Ainsi, sur 16 millions d’habitants, le nombre total d’hémophiles attendus serait de 3042 (46), alors que seuls 264 hémophiles sont répertoriés et suivis (21, 22).
Génétique
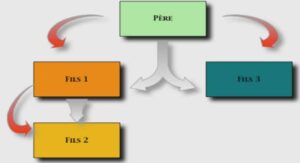
L’hémophilie est une maladie héréditaire, récessive, liée à une anomalie moléculaire des gènes des FVIII et FIX situés sur le chromosome X (40). Les gènes responsables de la synthèse des protéines des FVIII et FIX sont situés sur le bras long du chromosome X à deux endroits distincts : Xq28 pour le FVIII, Xq27 pour le FIX. Différentes anomalies génétiques à l’origine de l’hémophilie aboutissent à l’absence totale de FVIII ou de FIX fonctionnels et sont à l’origine de la plupart des formes sévères de la maladie. D’autres permettent la synthèse d’un taux faible de FVIII ou FIX circulant, à l’origine des formes modérées ou mineures. Dans une même famille, le type d’hémophilie (A ou B) et le degré de sévérité de la maladie sont toujours identiques (40). Chez les filles qui ont deux chromosomes X, l’anomalie du gène située sur un chromosome est en général compensée complètement ou partiellement par l’autre chromosome X sain. Elles ne seront pas malades mais conductrices de la maladie qu’elles pourront transmettre à leur descendance. Les garçons ayant un seul chromosome X ne peuvent pas compenser l’anomalie ; ils manifestent donc la maladie (35, 50). Dans 30% des cas, il n’existe pas d’antécédent d’hémophilie dans la famille, on parle alors d’hémophilie sporadique (40, 50). L’hémophilie féminine existe, mais elle est exceptionnelle. Le recours au conseil génétique est indispensable dès le diagnostic établi pour informer l’hémophile et sa famille et préparer à la prise en charge ultérieure de l’ensemble de la famille sur le plan génétique, en particulier pour aborder la transmission de la maladie (40). Ces conductrices peuvent transmettre l’anomalie génétique à leur descendance. Celle-ci va alors toucher 1 garcon sur 2 et 1 fille sur 2. Les enfants d’un garcon hémophile seront tous indemnes de la maladie s’ils sont de sexe masculin et leurs filles seront toutes des conductrices (35, 50) ; (Figure 1). 7 Figure 1 : Transmission de l’hémophilie (père hémophile, mère conductrice) I.4. Physiopathologie
Rôle du facteur VIII dans l’hémostase
Le facteur VIII de la coagulation circule dans le plasma sous forme liée à une proteine de transport, le facteur Von Willebrand (VWF). Son rôle est fondamental pour que le phénomène de coagulation puisse se dérouler de facon efficace. La coagulation est le deuxième temps de l’hémostase qui est un processus permettant de maintenir le sang à l’état fluide dans les vaisseaux. C’est un phénomène complexe déclenché dans l’organisme par l’activation du facteur VII au contact d’un récepteur cellulaire appelé facteur tissulaire. Le facteur VII devient alors le facteur VII activé qui active soit directement le facteur X en facteur X activé, soit le FIX en FIX activé. Le FIX activé seul est peu efficace, il a besoin pour augmenter son action de la présence du FVIII. Le complexe (FIX activé + FVIII activé) active lui aussi le facteur X en facteur X activé. La suite de la coagulation permet de générer un caillot de fibrine qui vient consolider le premier thrombus blanc formé au cours de l’hémostase primaire (figure 2). 8 Figure 2 : Schéma actuel de la coagulation I.4.2. Coagulation chez l’hémophile Le syndrome hémorragique est secondaire au défaut qualitatif ou quantitatif du FVIII ou FIX. Ces facteurs correspondent à une étape indispensable dans l’hémostase. Leur absence ou leur mauvais fonctionnement entraîne un caillot de mauvaise qualité à l’origine des saignements primaires ou de leur reprise. La sévérité du type d’hémophilie dépend du taux du facteur anti-hémophilique (FAH). Il existe en général une corrélation entre le taux circulant de FAH et les manifestations hémorragiques. Cependant le fait qu’il existe deux voies, d’une part une voie directe d’activation du facteur X par le facteur VII et d’autre part une voie indirecte passant par le complexe anti hémophilique, explique pourquoi l’hémophile ne saigne pas en permanence : la coagulation reste possible (35, 40).
Diagnostic
Diagnostic positif
Diagnostic clinique
Forme majeure
L’hémophilie se manifeste par des accidents hémorragiques, dont les plus fréquents intéressent l’appareil locomoteur (muscles, articulations, os) (40). A. Hémorragies non extériorisés Hémarthroses (35, 74) Les hémarthroses sont des épanchements sanguins intra-articulaires. Ce sont les accidents hémorragiques les plus fréquents chez l’enfant hémophile. Les premières hémarthroses apparaissent vers l’âge de 2 à 4 ans lors de l’apprentissage de la marche pour des traumatismes minimes ou de manière spontanée. Les articulations les plus atteintes sont les genoux, coudes et chevilles suivies en fréquence par les épaules et les hanches. Les autres articulations (poignets, mains, pieds) sont plus rarement affectées. o Hémarthroses aigues Elles se caractérisent par : Une douleur brutale, vive et permenante exacerbée par la mobilisation et la palpation ; elle est calmée en quelques heures par l’administration de FAH en dose efficace ; Une tuméfaction articulaire ; Une augmentation de la chaleur locale ; Une limitation de la mobilité et une attitude antalgique en flexion entrainant parfois une raideur musculaire. Des signes annonciateurs sont souvent rapportés par les patients (picotements notamment). La mesure du flexum permet le suivi évolutif (Figure 3). Figure 3 : Hémarthrose du genou droit 10 o Arthropathie subaigue L’arthropathie subaiguë débute après au moins deux épisodes aigus, lorsque se développent un épaississement des tissus mous péri-articulaires, une limitation des mouvements d’origine cartilagineuse et une atrophie musculaire secondaire à l’impotence fonctionnelle. o Arthropathie chronique Elle est fréquente et secondaire à la récidive des hémarthroses. Elle se traduit par une déformation et une limitation de l’articulation ainsi que par une diminution de la force musculaire. C’est une complication très invalidante à long terme d’où l’intérêt d’un traitement précoce des hémarthroses. Hématomes (3, 35) Les hématomes sont des hémorragies développées à l’intérieur des muscles. Ils peuvent se manifester dans tous les muscles spontanément ou à l’occasion de traumatismes minimes. Le muscle devient douloureux et gonflé (figure 4). Leur gravité est liée soit à leur volume (risque de spoliation sanguine), soit à leur localisation. Moins fréquents que les hémarthroses post-traumatiques dans 50% des cas, les hématomes musculaires se prêtent à plusieurs classifications : suivant l’accessibilité des muscles touchés, on oppose les hématomes superficiels (muscles des membres ou de la paroi abdominale) aux hématomes profonds (muscle psoas-iliaque) ; suivant le type de risque encouru : Il y a, d’une part, les hématomes faisant courir un risque vital par anémie aiguë, notamment chez le jeune enfant, par déperdition sanguine dans un muscle à grande gaine lâche (fessiers, cuisses, paroi abdominale) ou par obstruction ou compression des voies respiratoires et des axes vasculaires à destinée céphalique (langue, plancher de la bouche, cou). Et, d’autre part, les hématomes à haut risque fonctionnel dans les muscles à petite gaine serrée, comme la jambe (syndrome de loge), l’avant-bras et la main (syndrome de Volkmann), ou de par leur localisation à l’orbite (cécité), au creux axillaire, au pli du coude, à l’aine ou au mollet. Un hématome mal résorbé ou insuffisamment traité peut également évoluer vers une pseudotumeur hémophilique avec formation d’une coque fibreuse, adhérente aux plans adjacents qui peut ultérieurement évoluer pour son propre compte.
INTRODUCTION |