Télécharger le fichier original (Mémoire de fin d’études)
La stérilisation par la chaleur
Chaque cycle de stérilisation par la chaleur doit être enregistré avec l’exactitude et la précision adaptées par un appareil approprié, par exemple sur un graphique temps/température avec une échelle suffisante. La température doit être enregistrée par une sonde positionnée dans la partie la plus froide de la charge ou de la chambre chargée ; ce point ayant été déterminé lors de la validation ; la température doit de préférence être vérifiée à l’aide d’une seconde sonde de température indépendante située au même endroit. Le graphique ou sa photocopie doit faire partie du dossier de lot. Des indicateurs chimiques ou biologiques peuvent aussi être utilisés mais ils ne doivent pas remplacer les contrôles physiques (18) (19).
La stérilisation par la chaleur humide
Principe et dispositif technique dans une enceinte étanche appelée Autoclave, le produit à stériliser est soumis à de la vapeur d’eau surchauffée, ou à ruissellement d’eau surchauffée, à température élevée, sous une pression contrôlée, pendant un temps t. La stérilisation par la chaleur humide doit concerner uniquement des solutions aqueuses et des produits mouillés par l’eau. La température et la pression doivent être utilisées ensemble pour surveiller le procédé. L’enregistreur de température doit normalement être indépendant du régulateur et il doit exister un afficheur de température indépendant dont la lecture peut être régulièrement comparée à celle de l’enregistrement graphique durant la phase de stérilisation. Pour les stérilisateurs munis d’un drain dans la partie basse de la chambre, il peut aussi être nécessaire d’enregistrer la température à cet endroit tout au long de la phase de stérilisation. Des tests d’étanchéité de la chambre doivent être régulièrement effectués lorsque le cycle comprend une phase de vide. Des dispositions doivent être prises pour garantir que la vapeur utilisée pour la stérilisation est de qualité convenable et qu’elle ne contient pas d’additifs en quantités telles qu’ils contamineraient le produit ou l’équipement (18) (19).
La stérilisation par la chaleur sèche
Principe et dispositif technique dans une enceinte étanche appelée Four, le produit à stériliser est soumis à de l’air filtré sur filtre HEPA (High Efficiency Particulate Air) (figure 9) et chauffé (9). La stérilisation par la chaleur sèche peut être applicable aux liquides non aqueux et aux poudres. Le procédé utilisé doit comporter une circulation d’air dans la chambre et maintenir une surpression pour empêcher l’entrée d’air non stérile. En cas d’admission d’air, celui-ci doit passer à travers un filtre antimicrobien, par exemple un filtre HEPA. Lorsque le procédé de stérilisation par la chaleur sèche est aussi conçu pour détruire les pyrogènes, sa validation peut exiger l’utilisation d’endotoxines (18) (19).
La stérilisation par irradiation
Elle peut-être effectuer sur un médicament dans son emballage définitif (9). La stérilisation par irradiation est utilisée principalement pour la stérilisation de matières et de produits sensibles à la chaleur. De nombreux produits pharmaceutiques et certains articles de conditionnement étant sensibles aux radiations, cette méthode est acceptable uniquement lorsque l’absence d’effets nuisibles a été confirmée expérimentalement. Le rayonnement ultra-violet n’est pas une méthode acceptable de stérilisation finale. La dose d’irradiation doit être mesurée pendant le processus de stérilisation. A cette fin, les dosimètres utilisés doivent mesurer la dose réellement reçue par le produit, indépendamment de la dose d’irradiation délivrée. Les dosimètres doivent être placés dans la charge en nombre suffisant et suffisamment rapprochés pour qu’il y ait toujours un dosimètre dans l’enceinte. Lorsque l’on utilise des dosimètres en plastique, ils doivent être employés dans les limites de validité de leur durée de validité et d’étalonnage. Les dosimètres doivent être lus peu de temps après leur exposition aux radiations.
La stérilisation à l’oxyde d’éthylène
La stérilisation à l’oxyde d’éthylène (ou par tout autre agent stérilisant gazeux) nécessite une installation sophistiquée [8].Cette méthode de stérilisation ne doit être employée pour des produits qu’en l’absence d’alternative convenable. Différents gaz et agents fumigants peuvent être utilisés pour la stérilisation par exemple l’oxyde d’éthylène ou la vapeur de peroxyde d’hydrogène. L’oxyde d’éthylène doit être employé uniquement quand aucune autre méthode n’est utilisable (18). La figure 10 montre les différents stades auxquels peut se faire la stérilisation.
Table des matières
Liste des abréviations
Liste des figures
Liste des tableaux
Table des Matières
INTRODUCTION
Première partie : Généralités sur les formes pharmaceutiques administrées voie parentérale
I. Définitions
I.1 La voie parentérale
I.1.1 La voie intraveineuse (IV)
I.1.2 La voie sous-cutanée (SC)
I.1.3 La voie intramusculaire (IM)
I.1.4 Les autres voies d’administration moins courante
I.2 Le médicament
I.2.1 La définition juridique
I.2.2 La définition technologique
I.3 La forme injectable
I.3.1 Les préparations injectables
I.3.2 Les préparations pour perfusion
I.3.3 Les préparations à diluer pour injection ou pour perfusion
I.3.4 Les poudres pour injection ou pour perfusion
I.3.5 Les gels injectables
I.3.6 Les implants
I.4 La forme stérile
II. Les formes pharmaceutiques administrées par voie parentérale
II.1 Solution injectable
II.2 Suspension injectable
II.3 Emulsion injectable
II.4 Préparations à reconstituer
II.4.1 Poudres à reconstituer
II.4.2 Implants
II.5 Solvants
II.5.1 L’eau pour préparation injectable en vrac
II.5.2 L’eau stérilisée pour préparations injectables
II.5.3 La distillation à simple effet
II.5.4 La distillation à multiples effets
II.5.5 La technique de distillation par thermo-compression
III. La fabrication
III.1 Les différentes classes pharmaceutiques
III.2 Les différentes opérations de préparation
III.2.1 Classe A
III.2.2 Classe B
III.2.3 Classe C/D
III.3 Le comportement du personnel dans les différentes zones de ZAC
III.3.1 Classe A/B
III.3.2 Classe C
III.3.3 Classe D
III.4 La préparation
III.4.1 La préparation des solutions
III.4.2 La préparation des poudres
III.4.3 La préparation des émulsions
III.4.4 La préparation des suspensions
III.5 La répartition
III.6 La filtration stérilisante
III.6.1 La stérilisation par la chaleur
III.6.1.1 La stérilisation par la chaleur humide
III.6.1.2 La stérilisation par la chaleur sèche
III.6.2 La stérilisation par irradiation
III.6.3 La stérilisation à l’oxyde d’éthylène
III.7 Le mirage
III.8 L’impression ou l’étiquetage éventuel des ampoules
III.9 Le conditionnement
IV. Les caractéristiques
IV.1 La limpidité
IV.1.1 L’origine des particules
IV.1.2 Les méthodes de contrôle
IV.1.2.1 Les particules visibles
IV.1.2.2 Le contrôle de la contamination particulaire des solutions injectables pour perfusion (particules non visibles)
IV.1.2.2.1Les méthodes optiques automatiques
IV.1.2.2.2La méthode au microscope
IV.2 La neutralité
IV.2.1 La tolérance de l’organisme aux variations du pH
IV.2.1.1 Les préparations injectables non tamponnées
IV.2.1.2 Les préparations injectables tamponnées
IV.2.2 L’ajustement du pH
IV.2.3 Les contrôles
IV.2.3.1 La mesure du pH par les méthodes classiques
IV.2.3.2 La mesure du pouvoir tampon
IV.2.3.3 Les essais de conservation à différentes températures en fonction du pH et des agents utilisés pour l’ajustement du pH
IV.3 L’isotonicité
IV.3.1 La méthode d’étude hémolytique
IV.3.2 La méthode à l’hématocrite
IV.4 L’apyrogénicité
IV.4.1 L’origine et nature
IV.4.2 Les procédés d’élimination des pyrogènes
IV.4.2.1 L’adsorption sur charbon actif
IV.4.2.2 Le traitement par les oxydants
IV.4.2.3 La filtration
IV.4.2.4 Le chauffage en milieu acide ou alcalin
IV.5 La stérilité
V. ANALYSE DE RISQUES
V.1 Généralités
V.1.1 La définition de l’analyse de risques
V.1.1.1 L’approche déterministe
V.1.1.2 L’approche probabiliste
V.1.2 L’origine du mot risque
V.1.3 Qu’est-ce qu’un risque ?
V.1.4 La méthodologie générale
V.2 Les méthodes d’évaluation des risques
V.2.1 La classification des risques
V.2.2 La typologie des risques
V.2.2.1 Les démarches inductive et déductive
V.2.2.1.1La démarche inductive
V.2.2.1.2La démarche déductive
V.2.2.2 Les méthodes qualitatives et quantitatives
V.2.2.2.1Les méthodes qualitatives
V.2.2.2.2Les méthodes quantitatives
V.2.3 La gestion des risques
Deuxième partie : Etude expérimentale
I. CONTEXTE
II. CADRE D’ETUDE
II.1 Historique de l’usine
II.2 Présentation de l’usine
III. OBJECTIF DE L’ETUDE
III.1 Objectif général
III.2 Objectifs spécifiques
IV. MATERIEL ET METHODES POUR LA FABRICATION DU PRODUIT X INJECTABLE
IV.1 Matériel
IV.1.1 Le Thermo-compresseur
IV.1.2 La cuve « CIVINOX® »
IV.1.3 Le Skid de filtration
IV.1.4 Le Four « VISMARA® »
IV.1.5 La remplisseuse « BAUSCH STROBEL® »
IV.1.6 L’Autoclave « LEQUEUX ®»
IV.1.7 La mireuse « Eisai® AIM-287 »
IV.2 Méthodes
IV.2.1 Les fournisseurs
IV.2.2 La réception des matières premières et des articles de conditionnement
IV.2.3 La quarantaine
IV.2.4 Les magasins généraux
IV.2.5 La pesée
IV.2.6 La fabrication proprement dite du Produit X injectable
IV.2.6.1 La présentation du produit
IV.2.6.2 La sélection des lots
IV.2.6.3 La préparation de la solution
IV.2.6.3.1 Le contrôle physico-chimique et microbiologique de 900mL d’EPPI avant la préparation de la solution
IV.2.6.3.2La dissolution des Matières premières
IV.2.6.3.3Les paramètres préliminaires de la solution
IV.2.6.4 La décontamination des ampoules vides
IV.2.6.5 Le remplissage des ampoules
IV.2.6.5.1Les milieux de culture
IV.2.6.5.2Le contrôle des filtres
IV.2.6.5.3Le contrôle volumétrique des ampoules
IV.2.6.6 La stérilisation des ampoules remplies
IV.2.6.7 La chaine de conditionnement secondaire
IV.2.6.7.1Le triage
IV.2.6.7.2Le mirage manuel
IV.2.6.7.3Le mirage automatique
IV.2.6.7.3.1Les paramètres de l’appareil
IV.2.6.7.3.2Le « Knapp test »
IV.2.6.7.4Le contrôle des paramètres du mirage
IV.2.6.7.5Le conditionnement secondaire
V. RESULTATS
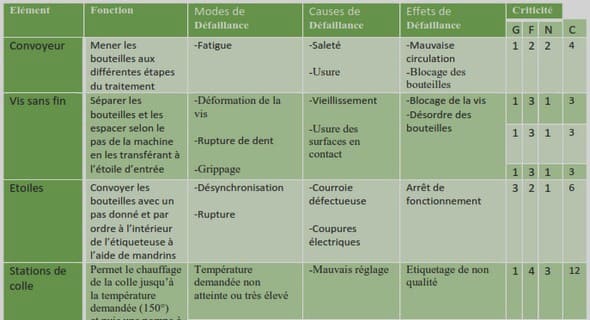
V.1 La définition de l’AMDEC
V.2 La gestion des risques
V.2.1 L’analyse des défaillances
V.2.2 L’évaluation des risques
V.2.3 Les recommandations pour la réduction des risques identifiés
V.2.3.1 La préparation des solutions
V.2.3.2 La décontamination des ampoules vides
V.2.3.3 Le remplissage des ampoules
V.2.3.4 La stérilisation des ampoules remplies
V.2.3.5 Le mirage des ampoules remplies et stérilisées
V.2.3.6 Le conditionnement des remplies mirées
VI. COMMENTAIRE
CONCLUSION
VII. REFERENCES