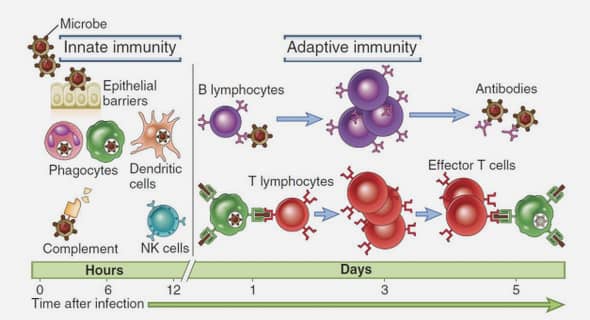
Etude du réarrangement propynyloxypyridines par catalyse électrophile et synthèse d’hétérocycles inédits
années 2000107, peut être étendue à d’autres groupements situés en position propargylique comme des phosphates ou des carbonates par exemple. Ces transformations permettent de développer des réactions complexes à partir de motifs facilement accessibles. Ce chapitre présente une étude de la réactivité des groupements hydroxypyridines propargyliques en catalyse électrophile. Différents réarrangements de ces motifs seront mis en évidence. Il sera montré que les conditions de réactions peuvent favoriser, voire rendre sélectif, l’un ou l’autre des chemins réactionnels possibles. Diverses structures hétérocycliques pourront être synthétisées grâce à ces méthodologies. Dans cette partie, nous présenterons le principe des réarrangements d’esters propargyliques catalysés par des métaux électrophiles. Cette présentation vise à introduire nos travaux sur la réactivité des 2-propynyloxypyridines en s’appuyant sur les études de mécanismes de réarrangements d’esters propargyliques. Ces propos seront illustrés d’exemples récents d’applications de ces réarrangements. Lorsqu’un ester propargylique est activé par un complexe métallique électrophile, le complexe métallique 2.1 qui en résulte peut se réarranger en suivant deux chemins réactionnels distincts : les voies a ou b (Figure 2-2). Dans le cas de la voie a, le complexe métallique subit une cyclisation de type 5-exo-dig du motif carbonyle de l’ester sur l’alcyne. La rupture hétérolytique de la liaison C-O de l’ester original dans l’intermédiaire 2.2 permet la formation de l’intermédiaire 2.3 de type carbène d’or. Formellement, cette réaction découle de la migration 1,2 du groupement acyloxy du substrat de départ. L’autre chemin réactionnel envisageable, la voie b, repose sur la cyclisation de type 6-endo-dig du motif carbonyle de l’ester sur la triple liaison activée. L’intermédiaire cyclique de type oxocarbénium 2.4 peut s’ouvrir pour former un allène 2.5. Cette transformation correspond à un réarrangement sigmatropique 3,3 de l’ester propargylique en carboxyallène 2 .5.
d’ester propargylique catalysée par le zinc avait été décrite par Ohloff en 1976.108 Le produit de la réaction est obtenu avec un très mauvais rendement. Presque dix ans plus tard, Rautenstrauch étudie une réaction similaire dans une réaction pallado-catalysée (Figure 2-3).109 La plus grande électrophilie des complexes de palladium(II) permet d’améliorer les résultats originaux obtenus par Ohloff et de préparer des cyclopenténones à partir d’éthynyl propényl acétates. Afin de rationaliser la formation des produits observés, deux chemins réactionnels qui reposent tous deux sur la migration 1,2 du groupement acyloxy de l’ester propargyliques ont été proposés. en 1950.110 L’utilisation de sels d’argent a ensuite permis de rendre cette méthode de synthèse d’esters alléniques plus efficace.111 Dès 1968, cette réaction a été appliquée par Benn à la synthèse de stéroïdes (Figure 2-4).112 Les allènes intermédiaires permettent de synthétiser des aldéhydes α−β-insaturés. Toutefois, ces méthodes qui reposent sur l’utilisation de sels de cuivre ou d’argent restent limitées en efficacité. En effet, l’ester propargylique et l’ester allénique correspondant sont présents en équilibre dans le milieu réactionnel. En conséquence, des mélanges, souvent inséparables, de ces deux espèces sont obtenus. Ces réactions ont donc été peu exploitées en chimie de synthèse jusqu’à ce que se développe la catalyse homogène à l’or.
platine(II) inspirée des travaux de Rautenstrauch.113 En accord avec le mécanisme postulé par Rautenstrauch, les auteurs proposent un mécanisme pour cette réaction qui repose sur la migration 1,2 du groupement ester catalysé par le platine(II). L’alcyne activé subit une cyclisation en mode 5-exo-dig. L’ouverture du cycle ainsi formé fournit un intermédiaire carbénique qui est ensuite piégé de manière intramoIéculaire par l’alcène nucléophile pour former un cyclopropyle. A la fin de leur travail, ils se sont demandé si d’autres métaux de transition hautement électrophiles ne pouvaient pas engendrer le même type de rearrangement dans des conditions plus douces. Ils ont montré que des complexes d’or(I) isolobaux au platine(II) générés in situ, catalysaient la même réaction à température ambiante de manière plus efficace que les complexes de platine(II). Les auteurs ont suggéré que l’étape-clé de cette transformation serait la formation d’un carbène d’or intermédiaire analogue à celui décrit dans le cas de la catalyse par un sel de platine.