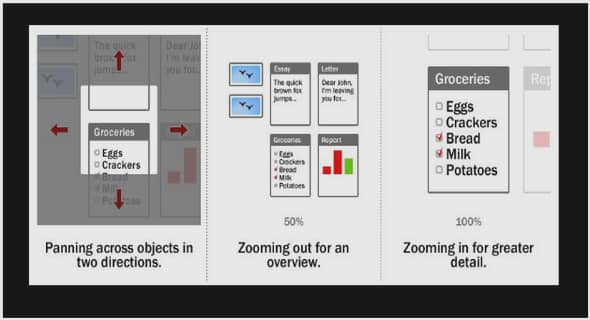
Modélisation de la ségrégation des phases liquides dans le système binaire U-O
Le système thermodynamique U-O
Le système binaire uranium-oxygène (U-O) est d’intérêt pour notre problématique puisqu’il s’agit du système thermodynamique le plus simple qui présente une lacune de miscibilité à l’état liquide tout comme le système d’intérêt U-O-Zr-acier pour la problématique de la stratification d’un bain de corium en cuve. A l’équilibre thermodynamique, deux phases liquides immiscibles (oxyde et métallique) coexistent dans un domaine de température et de composition [104]. La Figure 3.1 présente le diagramme de phase du système binaire U-O et met en évidence l’existence de cette lacune de miscibilité. Dans le modèle thermodynamique de la base NUCLEA [18, 49], la phase liquide est décrite par un modèle associé tel que discuté dans le chapitre 2. Les espèces du système sont l’uranium métallique U, le dioxyde d’uranium UO2 et l’oxygène « libre » O.La particularité de ce modèle associé réside dans la dépendance de l’énergie de Gibbs de la phase liquide aux fractions molaires des espèces associées. Dans les systèmes binaires du type A-B où les éléments A et B n’interagissent pas, et donc lorsqu’il n’y a pas de formation d’espèces associées AaBb, la seule connaissance d’une des deux fractions molaires (xA ou xB) est suffisante pour déterminer la composition du système. Ici ce n’est pas le cas comme le montre l’ équation (3.9), puisque la seule connaissance de la composition de l’oxygène xO ne permet pas de déterminer la composition locale (en termes d’espèces) du système, car l’oxygène au sens de l’élément se « partage » entre l’oxygène « libre » et le dioxyde d’uranium. Cela introduit notamment une complexité supplémentaire vis-à-vis des travaux menés par [85] sur la lacune de miscibilité liquide dans le système Bi-Zn puisque dans ce travail l’énergie de Gibbs de la phase liquide dépend uniquement des fractions molaires du bismuth (Bi) et du zinc (Zn). Ainsi, la seule connaissance d’une des deux fractions molaires s’avère suffisante pour décrire l’état local du système Bi-Zn. Un calcul de l’équilibre thermochimique du système binaire U-O est réalisé avec le minimiseur d’énergie de Gibbs Open CALPHAD [105]. Les résultats de ce calcul d’équilibre associé à la lacune de miscibilité du système binaire U-O sont présentés dans le Tableau 3.1.
Modèle de Cahn-Hilliard
Couplage avec la donnée thermodynamique
CALPHAD Nous avons discuté précédemment du choix de la composition locale en oxygène xO pour décrire l’état local du système binaire U-O. A température et pression constantes, l’énergie libre du système est alors décrite par la fonctionnelle de Ginzburg-Landau F de la forme suivante F = Z V ag˜ liq (xO) + κ 2 k∇xOk 2 dV (3.16) où g˜ liq est la contribution « homogène » de la fonctionnelle F, plus particulièrement il s’agit de la densité d’énergie libre de Gibbs définie telle que g˜ liq (xO) = G˜liq (xO) Vm (3.17) Vm étant le volume molaire et nous faisons l’hypothèse que celui-ci est constant dans notre modélisation, ce qui revient à considérer qu’il est indépendant de la composition. Cette hypothèse sera discutée plus en détail dans le chapitre 5 dédié au développement d’une modélisation cinétique sur le système d’intérêt U-O-Zr-Fe. κ est le coefficient associé au terme de gradient k∇xOk 2 . Il convient de noter ici que la forme de la fonctionnelle de Ginzburg-Landau diffère de la généralisation présentée dans le chapitre 2 par l’ajout d’un pré-facteur devant la densité d’énergie libre « homogène ». Comme discuté dans le chapitre 2, ce paramètre permet de traiter des épaisseurs d’interface supérieures à l’échelle de longueur caractéristique des interfaces physiques. Nous discuterons plus en détail ce point de modélisation dans la section 3.2.2.
Paramètres du modèle de Cahn-Hilliard
Dans cette partie nous abordons la détermination des paramètres de l’équation de Cahn-Hilliard binaire. Dans ce modèle cinétique, il y a deux types de paramètres. D’une part, les paramètres κ et a qui sont liés aux propriétés macroscopiques de l’interface : tension interfaciale et épaisseur de l’interface. D’autre part, le paramètre cinétique de mobilité chimique MO discuté notamment dans le chapitre 2. Intéressons nous aux paramètres qui déterminent les propriétés de l’interface. La tension interfaciale σ est une grandeur physique représentant l’excès d’énergie libre pour créer une interface, sa valeur peut être obtenue expérimentalement et devra être reproduite par notre modèle à interface diffuse. Comme discuté dans le chapitre 2, l’épaisseur de l’interface devient un paramètre numérique du modèle fixé par le maillage du domaine spatial.