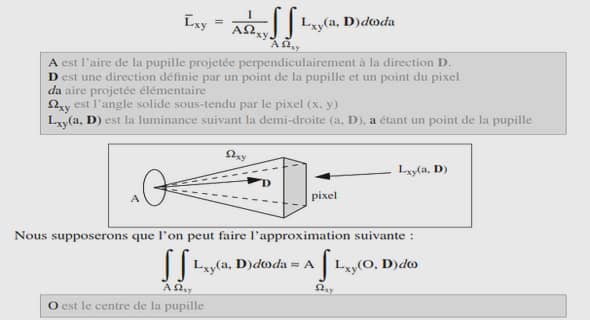
ATTITUDES THERAPEUTIQUES CHEZ LES DREPANOCYTAIRES A LA CLINIQUE INFANTILE
LES HEMOGLOBINES
L’hémoglobine est le constituant principal du globule rouge. Sa concentration moyenne ( CCMH ), est de l’ordre de 34g/dl, proche de la saturation. Son P.M est de 64.500. C’est la première grosse molécule parfaitement connue. Elle est formée de l’union dans le cytosol, de 4 groupements prosthétiques d’hème, pigment ferro-porphyrinique et de 4 chaînes polypeptidiques de globines identiques deux à deux : c’est un tétramère.
STRUCTURE DE L’HEMOGLOBINE
L’hème : (1)(2)(3) C’est une porphyrine contenant un atome de fer. La porphyrine ou protoporphyrine III comprend elle-même : – 4 noyaux pyrrols à sommet azoté réunis par des ponts méthènes ( -CH= ) – 8 chaînes latérales : méthyl, vynil ou acide propionique Le fer est au centre, fixé sur quatre (4) azotes des noyaux pyrrols et garde deux (2) valences libres. La molécule est plane.
La globine
Toutes les hémoglobines humaines comportent deux chaînes α couplées : – à deux chaînes β dans l’hémoglobine adulte majeure A ( HbA : α2β2 ) – à deux chaînes γ dans l’hémoglobine fœtale F ; d’ailleurs hétérogène pour le résidu 136γ Gly et γ Ala- ( HbF : α2γ2 ) – ou à deux chaînes δ dans l’hémoglobine adulte mineure A2 ( HbA2 : α2δ2 ). La chaîne α est constituée de 141 acides aminés ; les trois autres chaînes de 146 acides aminés, différents de 36 acides aminés entre β et γ ; de 10 entre β et δ. 28 La structure secondaire est en hélice discontinue à 8 segments avec des liaisons électrostatiques faibles entre acides aminés de spires voisines. La structure tertiaire est globulaire ménageant au centre une poche hydrophobe où s’insère l’hème La structure quaternaire est tétramèrique avec des contacts réduits entre les deux chaînes.
Liaison hème – globine
La structure tertiaire de chaque chaîne de globine ménage un repli superficiel appelé : » poche de l’hème » dans lequel se loge une molécule d’hème. L’arrimage se fait d’une part, par des liaisons qu’échangent les chaînes latérales acides propionique de l’hème et la globine ; d’autre part, par le fer qui dispose deux valences libres : l’une le fixe directement à la globine sur un résidu histidine dit » proximal » et l’autre intervenant sur la face opposée de la molécule d’hème fixe une molécule d’oxygène et par son intermédiaire assure un arrimage supplémentaire sur un autre résidu histidine dit » distal » de la globine.
Liaisons entre les 4 sous-unités
Les sous-unités constituées chacune d’une chaîne de globine portant sa molécule d’hème sont réunies entre elles par des nombreuses liaisons. (figure n° 1) Les liaisons α1β2 et α2β1 sont relativement peu nombreuses ( contact entre 19 acides aminés ) ; alors que les liaisons α1β1 et α2β2 sont plus fortes ( par 35 acides aminés ). α1 α2 β1 β2 Fer Hème Liaison Liaison forte Figure n° 1 : La molécule complète de l’hémoglobine
GENES DE LA GLOBINE
Chez l’homme normal, le gène α est dupliqué. Les deux gènes α sont présents sur le même chromosome (n° 16) à proximité l’un de l’autre ; et les gènes γ, δ, β situés, dans cet ordre, sur un autre chromosome (n° 11). Il n’y a qu’un seul gène β et un seul gène δ. En revanche, il y a deux gènes γ qui donnent naissance à deux chaînes qui ne diffèrent que par un seul acide aminé ( alanine ou glycocolle en 136ème position ) et qui ont la même migration électrophoretique. Il existe une certaine coordination dans la synthèse des gènes non α , et en cas de diminution de l’activité de l’un d’eux, on observe généralement une augmentation de l’activité de l’un ou de deux autres.
SYNTHESE DE L’HEMOGLOBINE
La biosynthèse de l’hémoglobine nécessite un équipement nucléaire complet qui n’existe que dans les cellules précurseurs des hématies. L’hématie humaine est en effet une cellule anucléée, donc dépourvue de l’équipement informationnel et enzymatique nécessaire à la synthèse des protéines. L’hémoglobine contenue dans les globules rouges a donc été synthétisée au cours des étapes des érythropoïèse. La biosynthèse de l’hémoglobine débute au stade de proérythroblaste et s’achève à celui de réticulocytes
Synthèse de l’hème
La synthèse de l’hème s’effectue indépendamment de celle de la globine. L’hème ne vient que secondairement s’accrocher aux chaînes polypeptidiques néosynthétisés pour réaliser la sous-unité d’hémoglobine. Certains étapes de sa synthèse sont localisés dans la mitochondrie, d’autre dans le cytosol. A partir de la glycine et de l’acide succinique, une série de précurseurs intermédiaires sont synthétisées : les porphyrines. L’importation du fer dans la protoporphyrine III réalise l’hème. La pyridoxine ( vitamine B6 ) est le coenzyme de l’Ala-synthétase et de l’hème synthétase aux deux extrémités de la chaîne de réaction qui mène à l’hème. 30 Le déficit de synthèse de l’hème n’est cependant qu’une conséquence très tardive et exceptionnelle de la carence en vitamine B6. La régulation de la synthèse de l’hème se fait essentiellement au niveau de l’Ala-synthétase, enzyme clef dont l’activité est inhibée par son produit direct l’Ala et par le produit final, l’hème. Glycine Acide succinique Ala – synthétase + B6 ALA (acide γ amino-lévulinique) Porphobilinogène Uroporphyrinogène Uroporphyrine (forme d’élimination) Coproporphyrinogène Coproporphyrine (forme d’élimination) Protoporphyrinogène Protoporphyrine III + Fer ++ HEME Figure n° 2 : Biosynthèse de l’hème (4) I – 4.2 Synthèse de la globine : (1)(4)(8) Elle se situe au niveau des polyribosomes des érythroblastes et s’achève dans le réticulocytes. Elle est codée par des gènes différents pour les 4 chaînes. Les deux paires des gènes α résultant d’une duplication sont situées sur le chromosome 16. Les gènes β et δ, proches l’une de l’autre, et le gène γ sont portés par le chromosome 11. 31 La synthèse des chaînes de globine est parfaitement régulée : il y autant de chaîne α que non α. Elle est activée, en outre, par l’hème lui-même déprimant sa propre synthèse par retro-inhibition de l’Ala-synthétase.
FONCTION DE L’HEMOGLOBINE
Pigment respiratoire des globules rouges. L’hémoglobine a une double fonction physiologique : – assurer le transport de l’oxygène (O2) des poumons aux tissus – et faciliter l’élimination des gaz carbonique (CO2) L’oxygénation tissulaire ne peut être assurée que si le transporteur peut se saturer en oxygène au niveau des poumons et le libérer efficacement dans un lit capillaire où sa pression partielle reste encore élevée. Ces exigences de la fonction oxyphorique ont pu être satisfaites par le fonctionnement allostérique de la molécule d’hémoglobine et l’effet régulateur des facteurs physico-chimiques environnementaux.
HEMOGLOBINE NORMALE
L’hémoglobine n’est pas la même à tout les âges. (figure n° 3) – L’embryon possède des hémoglobines particulières, GOWER et PORTLAND, associant des chaînes embryonnaires (figure ε ), fœtale ( figure γ ), et adulte ( figure α ), selon l’âge de l’embryon. – Chez le fœtus, l’hémoglobine F est le constituant hémoglobine principal de la vie fœtale. L’hémoglobine F a intrinsèquement une affinité pour l’oxygène identique à celle de l’HbA. – Chez l’adulte, on trouve simultanément plusieurs hémoglobines. Dans les semaines qui précèdent la naissance et celles qui la suivent, la synthèse de l’HbF est progressivement réprimée au profit de HbA qu’apparaît en fin de gestation, et de l’HbA2. Vers l’âge de six mois, on arrive à une formule voisine de celle de l’adulte. ( tableau n° 1 ).
INTRODUCTION |