Télécharger le fichier original (Mémoire de fin d’études)
Les éléments transposables chez les Eucaryotes
Histoire et définition
Le génome a longtemps été considéré comme étant relativement stable (aux mutations ponctuelles près) et évoluant lentement. Cette vision a été bouleversée dans les années 1940, par les travaux de Barbara McClintock portant sur des instabilités phénotypiques chez le maïs. Ces analyses ont montré que « des éléments de contrôle » Ac (Activateur) et Ds (Dissociation) sont capables de réguler l’expression des gènes responsables de la pigmentation des grains de l’épi (McClintock 1950; McClintock 1951). La découverte de ces séquences, a donc remplacé le paradigme statique par une théorie dynamique et fluide du génome dans lequel des réarrangements génétiques, parfois profonds, peuvent avoir lieu. Il a fallu attendre les années 1970, pour que l’intérêt porté à ces éléments se développe. A cette époque, Shapiro (1969) découvre que les éléments IS (séquences d’insertion) chez Escherichia coli sont impliqués dans l’acquisition d’une résistance à un antibiotique. Par ailleurs, chez Drosophila melanogaster, les éléments P et I ont été décrits comme étant responsables de la dysgénésie hybride (Kidwell et al. 1977, Bucheton et al. 1984). Ces séquences appelées aujourd’hui éléments transposables (ETs), sont capables de se répliquer et de se déplacer d’une position chromosomique à une autre dans un génome. Ces éléments bougent au sein d’une cellule, ce qui les différencie des virus. Ceci dit, de nombreux cas de transferts horizontaux entre espèces sont suspectés (Daniels et al. 1990; Loreto et al. 2008). La plupart des ETs possède des séquences promotrices et des séquences codantes nécessaires à leur transposition, même s’ils ont besoin de la machinerie de transcription et de traduction du génome hôte (comme les virus). Par ailleurs, les ETs sont de trois types (Figure 6) (Feschotte et Mouches 2000; Craig et al. 2002) :
– Les autonomes, qui contiennent un ou plusieurs gènes codant pour des protéines fonctionnelles impliquées dans leur mobilité et la régulation de leur expression.
– Les non-autonomes qui possèdent des cadres de lecture ouverts (ORFs) délétés ou mutés codant pour des protéines non fonctionnelles mais qui peuvent être mobilisés en trans par la machinerie d’un ETs autonome.
– Les éléments tronqués (mort) incapables de bouger.
Si sur le court terme, ces éléments peuvent avoir des effets délétères sur la structuration et le fonctionnement des génomes, sur le long terme, ils peuvent être source de variabilité génétique et peuvent également être domestiqués par l’hôte pour de nouvelles fonctions (exaptation). Ainsi, les ETs qu’on qualifiait de gènes égoïstes ou de parasites moléculaires sont considérés comme de véritables moteurs évolutifs d’adaptation et de plasticité génomique (Orgel et Crick 1980; Dawkins 1990; McDonald 1995; Britten 1996; Capy et al. 2000; Schmidt et Anderson 2006; Böhne et al. 2008).
Figure 6. Régulation en cis pour les éléments autonomes et en trans pour les éléments non-autonomes de Classe II (d’après Anxolabéhère et al. 2007).
Les éléments autonomes, codant une ou plusieurs enzymes, possèdent généralement des séquences terminales qui permettent la régulation en cis de leur transposition. Les éléments non – autonomes utilisent en trans les enzymes codées par les ETs autonomes.
Classification
Les éléments transposables ont été divisés, en fonction de leur mécanisme de transposition, en deux Classes : la Classe I ou rétrotransposons et la Classe II ou transposons à ADN (Figure 7) (Finnegan 1989,1992; Jurka et al. 2005; Wicker et al. 2007, Kapitonov et Jurka 2008) :
– Les Rétrotransposons ou Classe I transposent par l’intermédiaire d’un ARN transcrit par l’ARN polymérase II de l’hôte. Celui-ci est rétro-transcrit par la transcriptase réverse codée par le rétrotransposon, ensuite l’ADNc généré est inséré dans un nouveau locus du génome au niveau d’un site d’insertion TSD (terminal site duplication) spécifique par une intégrase. On parle ainsi d’une amplification réplicative selon le modèle copier-coller (Figure 8).
– Les transposons ou Classe II se déplacent via un intermédiaire ADN et sont subdivisés, selon leur mode de transposition conservatif ou réplicatif, en deux sous-classes (Figure 8).
Par ailleurs, en fonction de leur structure, de leur cycle de transposition, de leur domaine protéique ainsi que de leur site d’insertion TSD dans le génome, chaque Classe est subdivisée en différents ordres, superfamilles, familles qui peuvent coexister dans le même génome (Capy et al. 1997; Wicker et al. 2007).
Les éléments de la Classe I
Les éléments de la Classe I, ou rétrotransposons, peuvent être divisés en deux groupes : Les rétrotransposons à LTR qui possèdent de longues répétitions terminales (Long terminal repeat) et des rétrotransposons sans LTR.
Les rétrotransposons à LTR
Ils regroupent 5 grandes familles, Copia, Gypsy, Bel-Pao, les Rétrovirus et les rétrovirus endogènes (ERV). Leur taille varie de 5 à 10 kb (Bennetzen et al. 2005; Wicker et al. 2007; Charles et al. 2008). Ces ETs sont flanqués par deux répétions terminales identiques, longues et directes (LTR) dont la taille varie entre 100 pb à plusieurs kb.
Ces éléments autonomes contiennent principalement deux à trois ORF (Figure 7) :
– un gène gag codant pour trois protéines structurales de la capside virale chez les rétrovirus à savoir la protéine de la matrice et la protéine de la nucléocapside (Warmus et Brown 1989) ;
– un gène pol codant pour une polymérase nécessaire à la transposition constituée de quatre domaines : une protéase (PR), une transcriptase réverse (RT), une ribonucléase H (RH) et une intégrase (INT).
– un gène env, présent uniquement chez les superfamilles des rétrovirus et des ERV, codant pour la protéine d’enveloppe.
Il faut signaler que les éléments de l’ordre des DIRS (Dictyostelium Intermediate Repeat Sequences) dépourvus de LTR sont classés, de par la proximité phylogénétique de leurs séquences, avec les rétrotransposons à LTR (Poulter et Goodwin 2005). Ils présentent une tyrosine recombinase à la place de l’intégrase (Goodwin et Poulter 2001). Ces éléments sont subdivisés en trois superfamilles en fonction de leur structure terminale (Figure 7) : les DIRS1-like présentant des extrémités répétées inversées, les Ngaro et les VIPER présentant des répétitions directes dupliquées en 3’ mais qui différent par leur hôtes.
Figure 7. Classification des éléments transposables détectés chez les eucaryotes (d’aprés Wicker et al. 2007).
Les ETs de Classe I et II sont divisés en sous-classes, ordres et superfamilles. La taille du fragment dupliqué après insertion ou TSD (pour Target Site Duplication) est caractéristique de la plupart des superfamilles. Un code unifié à trois lettres a été proposé par ces auteurs :
– La première position correspond à la Classe : une transposition via un intermédiaire à ARN (R) ou transposition via un intermédiaire à ADN (D) ;
– La deuxième position correspond à l’ordre : par exemple T pour TIR répétitions terminales inversées ;
– La troisième position correspond à la superfamille : T pour Tc1-mariner, B pour piggyBac.
La distribution des éléments au sein des grands groupes est également donnée.
Quatre types de transposition sont résumés dans ce schéma. Les deux premiers opèrent selon un mode réplicatif. Ils correspondent à une transposition de type « copier-coller » et concerne les éléments de la Classe I. Le premier schéma (1) résume la transposition d’un rétrotransposon à LTR (la reverse transcription se fait dans la capside) et le deuxième (2) celle d’un rétrotransposon sans LTR pour lequel la reverse transcription se fait dans le noyau au moment de l’intégration. Les deux schémas suivants (3 et 4) concernent des éléments de la Classe II. Le schéma 3 représente un modèle de transposition de type « couper-coller » conservatif, relatif aux éléments de la sous-classe I. Au cours de ce cycle, le site cible, présenté en marron, subit une coupure à extrémités adhésives qui va conduire à sa duplication et la formation d’un TSD de part et d’autre de la copie de l’élément. Le schéma 4 correspond au mode de transposition des éléments de type Hélitron (sous-classe II) qui transposent selon un modèle « couper-coller » réplicatif. Un seul brin du transposon est clivé et transposé vers un nouveau site et la brèche laissée est réparée par des mécanismes de réparation de l’ADN en se basant sur la séquence du transposon original comme matrice. Au final un total de quatre copies est obtenu, soit deux par chromatide.
L’ADN génomique est représenté par une ligne droite noire, l’ETs en vert et l’ARN est par une ligne ondulée rouge. Les différents domaines protéiques sont représentés par des rectangles de couleurs : EN pour endonucléase, ENV pour enveloppe, INT pour intégrase, PR pour protéase, RH pour RNAseH, RT pour transcriptase inverse.
Les rétrotransposons sans LTR
Ces éléments se répartissent en deux grands groupes :
– les éléments autonomes qui incluent les LINE (Long Interspersed Nuclear Elements) et les PLE (Penelope-Like-Elements).
Les LINE ont une taille de 5 à 8 kb. Ils codent pour une transcriptase réverse et une endonucléase. Les différentes familles (R2, RTE, Jockey, L1 et I) possèdent une queue poly-A
à l’extrémité 3’et sont dépourvus d’intégrase mais possèdent une endonucléase, leur conférant un cycle de transposition différent de celui des rétrotransposons à LTR. Par ailleurs, ces éléments sont fréquemment tronqués en 5’ en raison d’intégrations imparfaites. Les éléments de la famille Penelope (PLE) présentent, quant à eux, des répétitions terminales inversées ou directes et possèdent un ORF unique de 2.5 kb qui code pour une transcriptase réverse et une endonucléase. De plus, ils peuvent comporter des introns (Evgen’ev et Arkhipova 2005).
– les éléments non-autonomes incluant les SINE (Short Interspersed Nuclear Elements). Les SINE ont une taille de 80 à 500 bp et ne possèdent ni ORF ni LTR. Néanmoins, ils comportent un promoteur d’ARN polymérase III en 5’ assurant leur transcription et une queue poly A qui est reconnue par la machinerie de transposition des LINE et notamment la RT ce qui les rend dépendant des LINEs (Unsal et Morgan 1995; Okada et al. 1997; Kramerov et Vassetzky 2005). L’élément Alu, trouvé dans le génome humain à plus de un million de copies, est responsable de plusieurs maladies (Deragon et Capy 2000 ; Roy-Engel et al. 2001; Dewannieux et al. 2003) est considéré comme étant le plus représentatif de cette superfamille.
Les éléments de la Classe II
Les éléments de la Classe II ou transposons à ADN sont subdivisés en deux sous-classes selon le mode de transposition conservatif ou réplicatif (Figure 7) (Feschotte et Pritham 2007; Wicker et al. 2007) :
– Les ETs de la sous-classe I sont coupés directement de l’ADN hôte puis insérés dans un nouveau site TSD, selon un modèle de transposition conservatif de type « couper-coller » assuré par un gène qui code pour la transposase (Figure 8).
– Les ETs de la sous-classe II transposent via un modèle réplicatif de type « copier-coller » ayant pour conséquence l’excision et le déplacement d’un seul brin d’ADN (Figure 8).
Par ailleurs, un groupe d’éléments non-autonomes MITE (Miniature Inverted Repeat Transposable Element) sont des versions délétées des transposons de Classe II et ne sont mobilisables qu’en trans par les ETs autonomes de cette Classe. Ils ont été décrits chez les plantes, les champignons, les amphibiens, les poissons et l’Homme (Bureau et Wessler 1994; Yeadon et Catcheside 1995; Izsvàk et al. 1999; Dufresne et al. 2007). Plusieurs mécanismes de réparation de l’ADN pourraient donner lieu à des délétions associées à des microhomologies au niveau des sites de rupture (Puchta 2005; McVey et Lee 2008). Le mécanisme abortive gap repair (AGR), proposé par Rubin et Levy (1997), est basé sur l’avortement de la synthèse de l’ADN, après excision d’un élément, à partir d’un brin complémentaire avant la fin de la réparation. Ceci conduit à l’émergence de diverses copies non autonomes ou à des copies délétées chimèriques rares avec une courte insertion interne (Brunet et al. 2002). D’autres mécanismes comme les systèmes non homologous end joining (NHEJ), microhomology – mediated end joining (MMEJ) et single strand annealing (SSA) peuvent également être impliqués. Ils sont capables d’induire des délétions de longueur variable à l’intérieur de divers éléments (Brunet et al. 2002; Negoua et al. 2013).
La sous-classe I : ordres des TIR et des Crypton
L’ordre des TIR, le plus fréquent, est caractérisé par des répétitions terminales inversées (TIR) qui régulent en cis la transposition et d’un ORF qui code une transposase. Il comprend 9 superfamilles : les Tc1-mariner (Plasterk et al. 1999), les hAT (Calvi et al. 1991), les Mutator (Lisch 2002), les P (Bingham et al. 1982), les Merlin (Feschotte 2004), les Transib (Kapitonov et Jurka 2003), les piggyBac (Fraser et al. 1983), les PIF-Harbinger (Jurka et Kapitonov 2001) et les CACTA (Kunze et Weil 2002). Une description plus détaillée des superfamilles Tc1-mariner et piggyBac, objet de notre recherche, sera présentée plus loin. Chaque superfamille est définie par sa structure, la longueur des TIR et les motifs du site catalytique de la transposase DDD/E responsable de l’excision et de l’intégration de l’élément (Plasterk et al. 1999; Feschotte et Pritham 2007; Wicker et al. 2007).
L’ordre des Crypton est représenté par une seule superfamille dépourvue de TIR et possède un ORF codant une Tyrosine recombinase à la place de la transposase, lui assurant un mécanisme de déplacement impliquant la recombinaison entre une molécule circulaire et l’ADN cible (Goodwin et al. 2003).
La sous-classe II : ordre des Helitron et des Maverick
Ces deux ordres sont représentés chacun par une seule superfamille.
La superfamille Helitron de taille de 5.5 à 17 kb est dépourvue de TIR et contient un ORF codant pour une tyrosine recombinase qui lui confère un modèle de réplication selon un mécanisme des cercles roulants (Kapitonov et Jurka 2001; Feschotte et Wessler 2001).
Les éléments Maverick (ou Politrons) sont, quant à eux, de longues séquences de 10 à 20 kb flanquées de TIR de 150 à 700 pb et codent jusqu’à 11 protéines, dont une polymérase, une intégrase de type rétrovirale, une protéase de type adénovirale et une ATPase, avec une variation de l’ordre des gènes selon les éléments (Kapitonov et Jurka 2006).
Caractéristiques, dynamique et impact des éléments transposables
En plus de leur capacité à se déplacer dans le génome, les ETs possèdent des caractéristiques qui peuvent leur conférer un rôle dans l’évolution des génomes.
Caractéristiques des ETs
Les éléments transposables sont ubiquitaires. Ils sont capables de se multiplier dans un génome mais également d’en coloniser d’autres via deux modes de transferts. Le premier mode correspond à un transfert vertical mendélien, ces éléments peuvent ainsi envahir le génome à partir d’un ancêtre, via la lignée germinale. Le deuxième mode, plus difficile à mettre en évidence, correspond au transfert horizontal (TH) d’ETs entre espèces reproductivement isolées et pouvant nécessiter l’intervention d’un organisme vecteur (navette). Généralement, les études illustrant les TH sont basées sur le pourcentage d’identité entre les séquences trouvées chez les espèces donatrices et réceptrices sans pour autant connaitre le vecteur (Pace et al. 2008; Loreto et al. 2008, Dupeyron et al. 2014; Tang et al. 2015). Ce mode de transfert a été en premier décrit avec l’élément P chez D. melanogaster, transféré horizontalement il y a 60 ans, à partir D. willistoni (Brookfield et al. 1984; Anxolabéhère et al. 1988; Daniels et al. 1990; Clark et al. 1994). Plusieurs arguments en faveur de ce mode de transfert ont été ensuite publiés. On peut citer ceux d’El Baidouri et al. (2014) qui ont montré la présence de rétrotransposons à LTR avec une distribution aléatoire chez 40 espèces de plantes phylogénétiquement éloignées appartenant aux Monocotylédones (i.e. Poaceae, Arecaceae, Musaceae) et aux Dicotylédones (i.e. Fabaceae, Vitaceae, Solanaceae…), et ceux de Gilbert et al. (2010) qui ont constaté que quatre familles distinctes de transposons chez Rhodnius prolixus, vecteur de la maladie de Chagas chez l’homme, ont été retrouvées chez deux hôtes de mammifères et que l’une de ces familles est présente chez des limnées, vecteurs de trématodes infectant divers vertébrés, et chez les mammifères de l’ancien monde.
Malgré leur présence dans les génomes, la répartition des ETs est variable d’une espèce à une autre indépendamment de la complexité (e.g. le nombre de fonctions cellulaires) de celui-ci (Figure 9) (Hua-Van et al. 2005; Feschotte et Pritham 2007). En effet, les ETs peuvent constituer la majeure partie d’un génome tels que ceux des poacées avec 90% chez le blé, 85% chez le maïs et 60% chez le riz (Goff et al. 2002; Schnabel et al. 2009; Biémont 2010). A l’inverse, chez l’abeille Apis mellifera (Biémont 2010) et la levure Saccharomyces cerevisiae (Spellman et al. 1998) les ETs ne représentent respectivement que 1% et 5% des génomes.
La proportion des ETs peut être quasiment identique pour un même groupe phylogénétique comme les primates (Homme, chimpanzé et macaque) avec 45%. Cependant, chez d’autres espèces proches, comme par exemple D. melanogaster et D. simulans, la proportion des ETs est variable, avec 15% et 5% respectivement (Clark et al. 2007).
Par ailleurs, au sein d’un même génome, la proportion des différentes familles d’ETs peut être très variable et certaines superfamilles ou Classe peuvent être plus abondantes (Feschotte et Pritham 2007; Pritham 2009), sans qu’aucune règle puisse être dégagée. Par exemple, les éléments de Classe I sont prépondérants dans les génomes des végétaux (e.g orge, blé) et dans ceux des mammifères (e.g. Homo sapiens) alors que les génomes des invertébrés (e.g. Acyrthosiphon pisum) présentent une majorité d’ETs de Classe II (Figure 10).
Dynamique des ETs dans les génomes
La dynamique ou cycle des ETs est généralement divisée en trois phases (Figure 11). La première correspond soit à l’invasion de la population suite à l’arrivée d’un nouvel élément actif par transfert horizontal dans le génome d’une espèce, soit à la réactivation d’une copie précédemment inactive (Kidwell 1992; Sanchez-Gracia et al. 2005). Dans une telle situation, un élément doit être très efficace de façon à se multiplier rapidement dans le génome hôte, sinon, il risque d’être éliminé par simple dérive génétique et/ou sélection (Le Rouzic et Capy 2005).
L’étape suivante est une phase durant laquelle des mécanismes de régulation ainsi que l’apparition de mutations et de délétions limitent l’invasion de l’élément. Ces mécanismes peuvent être de plusieurs types :
– régulation par la répression de la transcription comme observé pour les séquences d’insertions bactériennes IS (Zerbib et al. 1990).
– autorégulation par la surproduction de la transposase (overproduction inhibition OPI) entrainant la formation d’oligomères inactifs ou moins actifs qui diminuent l’efficacité du processus de transposition, ou par l’inhibition compétitive entre les éléments actifs et inactifs pour l’accès aux répétitions terminales inversées (TIRs) ce qui bloquerait l’excision des copies actives. Les copies délétées agissent comme des inhibiteurs négatifs dominants de la transposition (dominant negative complementation DNC). Ces deux modèles ont été décrits chez D. melanogaster avec l’élément mos1 de la famille mariner (Lohe et Hartl 1996; Lohe et al. 1997).
– régulation épigénétique par la méthylation d’ADN (Martienssen et Baron 1994; Slotkin et Martienssen 2007) ou par les ARN anti-sens formant des dimères avec les ARNm des ETs (Sijen et Plasterk 2003; Petit et al. 2007; Dowling et al. 2017).
Enfin, la phase de sénescence durant laquelle une dérive aléatoire peut entrainer une forte diminution du nombre de copies jusqu’à l’élimination complète de la famille. Ceci peut être dû soit à l’inactivation verticale par accumulation de mutations conduisant à la perte totale d’activité des ETs ainsi que l’impossibilité d’être mobilisé en trans (Lohe et al. 1995; Lohe et al. 1997; Le Rouzic et Capy 2005), soit à des recombinaisons ectopiques (Biémont et al. 1997; Kalendar et al. 2011) ou encore à la perte stochastique de copies par dérive génétique (Capy et al. 1992a; Lohe et al. 1995).
Cette figure résume la composition en ETs et la taille des génomes de différentes espèces. La distribution des ETs dans ces génomes semble stochastique. On note chez certaines espèces, une abondance d’ETs associée à une augmentation de taille du génome comme chez le maïs et le riz. Toutefois, on remarque aussi la faible proportion d’ETs (comme chez l’abeille), souvent associée à des phénomènes de contraction du génome et à la perte de grande quantité d’ADN.
Données obtenues à partir du séquençage de chacun de ces génomes : Apis mellifera [Honeybee Genome Sequencing Consortium, 2006], Arabidopsis thaliana [Kaul et al. 2000], Danio rerio [Haffter et al. 1996], Crocodylus porosus [St John et al. 2012], Drosophila simulans et D. melanogaster [Clark et al. 2007], Gallus gallus [Brandström et Ellegren, 2007 ], Homo sapiens [Lander et al. 2001], Macaca mulatta [Gibbs et al. 2007], Mus musculus [Chinwalla et al. 2002], Ornithorhynchus anatinus [Warren et al. 2008], Oryza sativa [Goff et al. 2002], Pan troglodytes [Chimpanzee Sequencing and Analysis Consortium, 2005], Saccharomyces cerevisae [Spellman et al. 1998], Takifugu rubripes [Kurowaka et al. 2005], Xenopus tropicalis [Hellsten et al. 2010], Zea mays [Schnable et al. 2009].
La figure montre les fractions de génomes occupées par les grandes familles d’ETs. Les variations de proportion des différentes familles d’ETs sont souvent liées à une amplification massive. Chez l’homme, les ETs sont dominés par des rétrotransposons sans LTR (LINE et SINE) suivi de rétrotransposons à LTR. La tendance est inversée chez les triticales (orge et blé) avec une dominance de rétrotransposons à LTR (e.g. Gypsy, Copia). Toutefois, dans le génome de l’orge et du puceron vert du pois, les éléments de Classe II sont bien plus abondants que chez H. sapiens.
Le groupe noté « autres » inclut des régions répétitives qui correspondent à des consensus d’ETs spécifiques à l’hôte mais qui n’ont pas pu être classées par le pipeline REPETs vu l’absence de caractéristiques structurelles et de similarités avec les ETs connus.
Données obtenues à partir du séquençage de chacun de ces génomes : Acyrthosiphon pisum [International Aphid Genomics Consortium 2010], Homo sapiens [Lander et al. 2001], Hordeum vulgare [Mazaheri et al. 2014], Triticum urartu [Daron et al. 2014].
Dans un même génome l’ensemble des ETs ne sont pas tous dans la même phase de leur cycle. Aussi, il peut y avoir de nombreuses différences d’une famille à une autre, certains ETs sont dans une phase d’invasion alors que d’autres sont dans leur phase de sénéscence (Le Rouzic et al. 2007).
Bien que la plupart des copies vont être perdues au cours du temps, certaines d’entre elles peuvent s’insérer dans des régions où elles induisent une augmentation de la fitness des individus qui les portent (insertions adaptatives). D’autres peuvent être domestiquées ou « exaptées » et survivent dans le génome hôte où elles sont « récupérées » pour une nouvelle fonction. Généralement, ces séquences sont en copie unique, elles sont conservées et persistent dans la population grâce aux effets de la sélection (Le Rouzic et al. 2007; Jurka et al. 2012).
Par ailleurs, elles sont responsables d’un balayage sélectif dans la région de leur insertion. Par exemple, des travaux antérieurs ont montré que la résistance à l’insecticide DDT chez D. melanogaster et D. simulans est corrélée à l’insertion du rétrotransposon Accord dans la région 5 ‘ du gène Cyp6g1 du cytochrome P450. Une réduction significative de la variabilité s’étendant au moins 20 kb en aval du gène de résistance a été observée (Catania et al. 2004; Schlenke et Begun 2004)., les levures, les protozoaires et les vertébrés (Yusa 2015).
Table des matières
Introduction Bibliographique
I. Les céréales : sous-famille des Pooideae
1. Systématique
2. Evolution des génomes
3. La céréaliculture
4. Les pathogènes et les ravageurs des céréales
4.1. Les agents pathogènes des céréales
4.1.1. Les virus
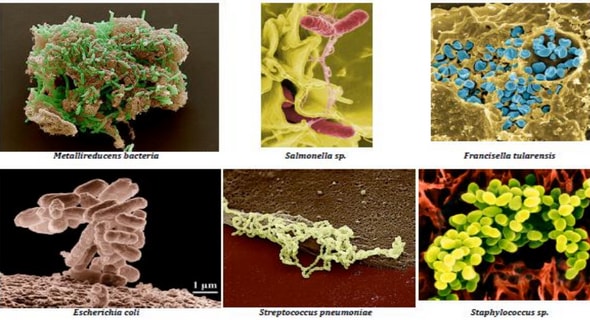
4.1.2. Les bactéries
4.1.3. Les champignons
4.2. Les ravageurs des céréales
4.2.1. Les nématodes
4.2.2. Les insectes
II. Les pucerons des céréales
1. Systématique
2. Caractéristiques chromosomiques
3. Cycle biologique
4. Dégâts des pucerons
5. Stratégies de lutte
III. Les éléments transposables chez les Eucaryotes
1. Histoire et définition
2. Classification
2.1. Les éléments de la Classe I
2.1.1 Les rétrotransposons à LTR
2.1.2 Les rétrotransposons sans LTR
2.2. Les éléments de la Classe II
2.2.1 La sous-classe I : ordres des TIR et des Crypton
2.2.2 La sous-classe II : ordre des Helitron et des Maverick
3. Caractéristiques, dynamique et impact des éléments transposables
3.1. Caractéristiques des ETs
3.2. Dynamique des ETs dans les génomes
3.3. Impacts des éléments transposables
3.4. Utilisation des éléments transposables en biotechnologie
IV. Les superfamilles Tc1-mariner et piggyBac
1. La superfamille Tc1-mariner
1.1. Caractéristiques générales des MLE
1.2. Structure des MLE
1.3. Mécanisme de transposition des MLE
2. La superfamille piggyBac
2.1. Structure des PBLE
2.2. Structure des séquences domestiquées PGBD
2.3. Mécanisme de transposition des PBLE
Délimitation du sujet
Matériel et Méthodes
I. Matériel biologique
II. Méthodes
1. Extraction de l’ADN
1.1. Méthode de Doyle et Doyle (1987)
1.2. Méthode basée sur l’utilisation des kits d’extraction
2. Amplification et purification de l’ADN
3. Clonage et purification des plasmides
3.1. Préparation des bactéries compétentes
3.2. Préparation du vecteur recombinant par ligation
3.3. Transformation et sélection des bactéries
4. Filtrage et vérification des séquences
5. Recherche in silico des éléments transposables dans les génomes
6. Alignement et traitement des séquences
7. Estimation de la contrainte sélective
8. Classification par la méthode agrégative UPGM-VM (Unweighted Pair Group Method with Variation Metric)
9. Construction des phylogénies moléculaires
Résultats
Chapitre I
Chapitre II
Chapitre III
Discussion générale & Perspectives
Références Bibliographiques