CONTRIBUTION A L’ETUDE D’IXAZOMIB NINLARO*
MEDICAMENT
FORMATION DES CELLULES CANCEREUSES DE MYELOME
Historique Le myélome multiple est reconnu depuis l’antiquité. Le premier cas bien documenté a été signalé en 1844 par Samuel Solly. Le cas le plus communément reconnu est celui de thomas Alexander McBean, un commerçant hautement respectable de Londres en 1850. L’un des cas les plus connu de 10 myélome multiple était celui du Docteur Loos qui a été rapporté par Otto Kahler. Le myélome multiple aussi connu sous le nom de maladie de kahler a été désigné par le médecin Otto kahler .
Caractéristiques du myélome multiple
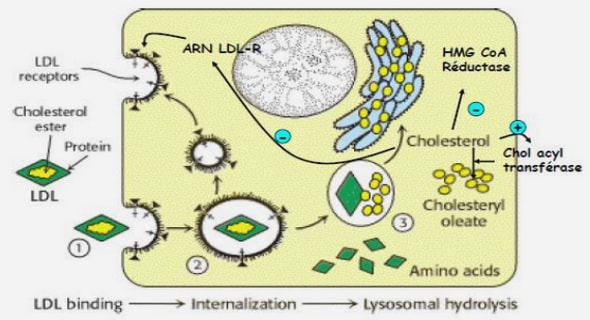
Le myélome multiple est une hémopathie maligne, aussi appelé cancer de la moelle osseuse. Il se caractérise par la prolifération excessive dans la moelle osseuse d’un type de globule blanc nommé plasmocyte, devenu anormal [8]. Le myélome multiple ou myélome plasmocytaire fait partie d’un groupe nosologique qui porte la dénomination de néoplasies plasmocytaires dans la classification OMS des hémopathies malignes lymphoïdes [6]. Ce groupe d’affections comprend également les plasmocytomes solitaires, osseux et extraosseux, les maladies avec dépôts d’immunoglobulines (Ig) monoclonales dont l’amyloïdose primaire (amyloïdose AL), et les maladies des chaînes lourdes. Figure 3 : Lésions osseuses lytiques du crâne [2]. Figure 4 : Infiltration plasmocytaire de la moelle hématopoïétique en cytologie [2]. Le myélome multiple est une affection caractérisée par l’expansion d’un clone lymphocytaire B à un stade terminal de la différenciation avec production d’une immunoglobuline monoclonale. Le myélome multiple comprend classiquement des lésions osseuses lytiques (fig. 3), une infiltration plasmocytaire de la moelle osseuse (fig. 4) et la présence d’une immunoglobuline monoclonale (paraprotéine), habituellement décelée dans le sang et/ou dans les urines. Les plasmocytes anormaux produisent, eux aussi, une immunoglobuline ou un fragment d’immunoglobuline, présente en quantité anormalement élevée et qui ne joue plus son rôle dans le cadre du système immunitaire. On l’appelle immunoglobuline monoclonale. Dans de très rares cas, il arrive toutefois que le myélome ne sécrète pas d’immunoglobuline lorsque la maladie n’engendre pas de signes cliniques ou biologiques [8]. Dans les cancers plasmocytaires, ces plasmocytes sont produits en trop grand nombre et sont incapables d’effectuer leur travail. Ces cellules prennent également la place des cellules sanguines normales (globules rouges, autres globules blancs, plaquettes). La diminution des cellules normales ainsi que l’accumulation des cellules anormales plasmocytaires en véritable tumeur intraosseuse sont à l’origine des symptômes de la maladie. De plus la production d’anticorps en trop grand nombre par ces cellules peut abîmer les reins et rendre le sang moins fluide [32]. 12 Signes En fonction de son évolution, la maladie peut provoquer un certain nombre de symptômes, tels que des douleurs osseuses ou des fractures, une insuffisance rénale, une anémie, une hypercalcémie [8]. Physiopathologie La différenciation du lymphocyte B (LB) normal comprend 4 grands stades corrélés avec le réarrangement des gènes d’immunoglobulines (Ig). La cellule pro-B (progéniteur B) présente dans la moelle osseuse subit des réarrangements des gènes du locus de la chaîne lourde des Ig (IgH pour « Heavy Ig ») : d’abord une recombinaison entre un segment D et J, puis un second réarrangement entre un segment V et le complexe DJ. Elle devient alors une cellule pré-B (précurseur B) qui exprime un pré-BCR (B cell receptor). Ces précurseurs préB prolifèrent, permettant d’expandre ce compartiment. Les cellules pré-B sont ensuite soumises à un réarrangement du locus (locus ou de la chaîne des Ig légères (IgL pour « Light Ig »), ce qui conduit à des cellules B immatures possédant un BCR fonctionnel sous forme d’IgM. Le stade de cellule B mature est caractérisé par une co-expression membranaire de l’IgM et IgD. Ces LB matures naïfs quittent la moelle osseuse et entrent dans la circulation périphérique jusqu’aux organes lymphoïdes pour poursuivre leur différenciation, après contact avec l’antigène (Ag). Dans les organes lymphoïdes secondaires, l’activation des LB de manière T-dépendante va déclencher leur division. Une partie de ces cellules B se différencie en plasmocytes sécrétant des anticorps IgM de faible affinité (plasmocytes à courte durée de vie). Certains lymphocytes B(LB) actifs vont après dans un follicule pour créer le centre germinatif. Les cellules seront soumises à des hypermutations somatiques au niveau des régions variables réarrangées de l’Ig, permettant de sélectionner les LB avec une forte affinité pour l’Ag. L’affinité de liaison du BCR pour l’Ag (stade centroblaste) 13 sera alors modifiée. Ce processus permet de sélectionner uniquement les cellules exprimant un BCR de forte affinité pour l’Ag (stade centrocyte). Les centrocytes sélectionnés de façon positive vont ensuite subir une commutation isotypique qui engendrera un changement de classe de l’IgM vers IgG, IgA ou IgE membranaire. Ces LB générés vont se différencier soit en LB mémoires soit en plasmablastes qui rejoindront la moelle osseuse ou les muqueuses où ils termineront leur différentiation en plasmocytes matures, synthétisant des Ig en grandes quantités (plasmocytes à longue durée de vie). Cette dernière étape de différentiation intramédullaire des plasmablastes en plasmocytes reste peu connue (Figure 5) [23]. Les cellules B précurseurs (pro-B puis pré-B), présentes dans la moelle osseuse, subissent des réarrangements des gènes du locus de la chaîne lourde des IgH (segments VDJ) et de ce/7ux de la chaine des IgL, ce qui conduit à des cellules B immatures. Les cellules exprimant un BCR fonctionnel de type IgM (cellules B matures) quittent la moelle osseuse et vont dans la circulation périphérique jusqu’aux organes lymphoïdes. Après contact avec un antigène, ces cellules vont soit se différencier en plasmocytes à courte durée de vie ou soit former un centre germinatif où seulement les lymphocytes B de forte affinité seront sélectionnés (mécanismes d’hypermutation et de sélection des antigènes). A l’issu de ce processus, les lymphocytes sélectionnés vont se différencier soit en lymphocytes B mémoires soit en plasmablastes qui rejoindront la moelle osseuse où ils termineront leur différentiation en plasmocytes à longue durée de vie [23] Les translocations chromosomiques en 14q32 sont caractéristiques des hémopathies malignes de la lignée B. Elles intéressent le locus codant pour la chaîne lourde des immunoglobulines (IgH). On pense que cette erreur de recombinaison du locus IgH survient dans 15 un lymphocyte B mémoire, activé dans les centres germinatifs ganglionnaires ou spléniques, lors de l’hypermutation somatique ou surtout de la commutation de classe [24]. Ces translocations vont aboutir à mettre les gènes partenaires sous la dépendance des enhancers du locus IgH, et donc à les hyperexprimer[24]. Au cours du myélome multiple, de multiples anomalies cytogénétiques s’accumulent. Elles peuvent être détectées par des techniques d’hybridation ¨ in situ¨ (FISH) sur des cellules non proliférantes .
INTRODUCTION |