Biochimie des protéines et des acides nucléiques
Culture de Saccharomyces cerevisiae et extraits de levure
Souches utilisées
Plusieurs souches de levure Saccharomyces cerevisiae ont été utilisées durant cette étude (tableau 2.1. pour les deux principales). Dénomination Génotype YDS 38 MATα, ade2-1, trp1-1, can1-100, leu2-3, 112, his3-11, 15, ura3-1, hmr :: TRP1 EG124 = GA59 MATa, trp1 ura3-52 leu2-3, 112 prb1-1122 pep4-3 prc1-407 gal2 Tableau 2.1. : Les deux principales souches de levure sources des extraits protéiques étudiés dans ce projet. Nous avons commencé par travailler avec la souche YDS 38. A partir de décembre 1998, tout le travail a été effectué avec la souche EG124 construite pour produire peu de protéases (Cockell et al, 1995).
Conditions de culture
La culture des levures Saccharomyces cerevisiae est réalisée en milieu liquide YPG (Yeast Peptone Glucose), sous agitation permanente (oxygénation des levures), à une température de 30°C. Pour permettre un bon démarrage de la culture, une colonie isolée provenant de levures souches (stockées sur boîtes de Petri à 4°C) est prélevée pour incubation pendant 24h dans un milieu de préculture d’une vingtaine de millilitres. Ce milieu est ensuite versé directement dans un fernbach de 1 litre de YPG pour 60 heures environ à 30°C sous agitation continue. La croissance des levures est suivie par la mesure de l’absorbance à 600 nm, lors de prélèvements de 1 ml du milieu de culture. Le développement de la culture est stoppé en fin de phase de croissance exponentielle.
Extraction des protéines
Plusieurs méthodes permettent de briser les levures en vue d’en extraire des protéines. Parmi les plus répandues, citons la méthode consistant à les écraser, dite « French press », ou celle consistant à déchirer les membranes par frottements contre des billes de verre. Nous avons commencé à travailler selon ces modes de préparation (cf. Annexes). Ces deux méthodes ne permettent pas de traiter des quantités conséquentes de levures (faible volume de la chambre d’écrasement dans le cas de la French press et viscosité croissante du milieu d’extraction dans le cas des billes de verre, diminuant d’autant l’efficacité de ces méthodes). Ne pouvant atteindre un objectif de production (plusieurs millilitres d’extraits de protéines à une concentration moyenne de 1 µg / 1µl, provenant de plusieurs dizaines de grammes de levures) nous permettant de travailler durablement avec des protéines venant d’une même préparation, nous avons développé une méthode d’extraction plus adaptée à nos besoins. Cette méthode repose sur la cassure des levures par des congélations quasi-instantanées (azote liquide) et successives, avec entre chaque congélation des périodes de réchauffement en bain-marie à 25°C – 30°C. Pour faciliter ces opérations, les levures récupérées par centrifugation après culture, sont remises en suspension dans un faible volume d’eau (maximum 5% volume / masse des levures). Nous avons pu contrôler l’efficacité de la méthode par inspection des cellules au microscope, lors des étapes de congélation/décongélation. En présence de SDS (1%), les levures cassées forment des agrégats alors que celles dont les membranes sont intactes restent isolées. La première congélation n’est efficace qu’à 30% alors qu’après la seconde congélation, plus de 95% des levures sont agrégées, donc effectivement cassées. Cependant, cette technique ne s’applique pas à la récupération de protéines membranaires, car les membranes sont simplement rompues. Elle ne donne pas non plus de résultats satisfaisants lorsque les levures sont collectées en phase stationnaire (membranes trop épaisses).
Protection contre les protéolyses
L’ensemble des étapes menant à l’obtention d’extraits de protéines de levure se fait en présence d’anti-protéases (voir modes opératoires en Annexes) afin d’éviter les protéolyses, principalement Chapitre II. Matériels et Méthodes – 22 – lors des phases de réchauffement. Les anti-protéases utilisées au cours de la préparation de nos extraits protéiques, ainsi que les concentrations efficaces sont données dans le tableau 2.2.. Anti-protéases Cibles Concentrations habituellement utilisées PMSF Trypsine, Chymotrypsine, protéases à sérine 0.1 – 1 mM Pefabloc Kallikrein, Thrombine, TPA, Subtilisine A, Trypsine, Chymotrypsine, Plasmine, protéases à sérine 0.4 – 4 mM Benzamidine Peptidases 0.1 µM Aprotinine Kallikrein, Trypsine, Chymotrypsine, Plasmine 0.01 – 0.3 µM Inhibiteur de Trypsine (ovomucoïde) Trypsine 0.5 – 5 µM EDTA Metalloprotéases 0.5 – 1.3 mM Tableau 2.2. : Anti-protéases utilisées lors des étapes d’extraction des protéines de levure et de purification par chromatographie liquide. (sources : Sambrook et al., 1989 ; Boehringer Mannheim : The complete guide for protease inhibition, 1998)
Préparation des extraits protéiques.
Dosage colorimétrique des protéines. Les levures cassées sont centrifugées et le surnageant récupéré. Les acides nucléiques présents sont éliminés par précipitation à la streptomycine (8mg /ml) (Spelsberg, 1983). Aucune amélioration à cette séparation n’est apportée par une précipitation complémentaire à l’aide de polyéthylèneimine (PEI) (Murray P., 1990). Nous avons vérifié qu’aucune co-précipitation importante de protéines du surnageant n’avait lieu lors de ces étapes en hydrolysant les acides nucléiques précipités à la streptomycine (utilisation d’une endonucléase, la benzonase, et gel de protéines). Les protéines sont ensuite précipitées au sulfate d’ammonium à 55%, et le précipité obtenu est repris dans du tampon Tris (45 mM) borate (45 mM) EDTA (1 mM) ( = TBE 0.5x) à pH 8, en présence de β-mercaptoéthanol (5mM), de détergent non ionique Nonidet P40 (20µl/l), de PMSF (0.5 mM), de Pefabloc (4 mM), de benzamidine (6 µM), d’aprotinine (8.5 nM), d’inhibiteur de trypsine (3 nM) et de NaCl (50 mM). L’extrait de protéines obtenu est dialysé pour éliminer le sulfate d’ammonium, pendant 5 heures à 4°C contre du TBE 0.5x en présence de NaCl 10 mM, βmercaptoéthanol 5mM, de MgCl2 2 mM et de Nonidet (20µl/l). Chapitre II. Matériels et Méthodes – 23 – Le dosage des protéines présentes dans l’extrait est réalisé par colorimétrie (Bradford, 1976) à l’aide du kit Biorad Protein Micro Assay (1-20µg/l)). Une gamme étalon est réalisée à l’aide de BSA (sérumalbumine bovine). La concentration finale en protéines des extraits préparés varie de 1 à 2 µg protéines /µl d’extraits.
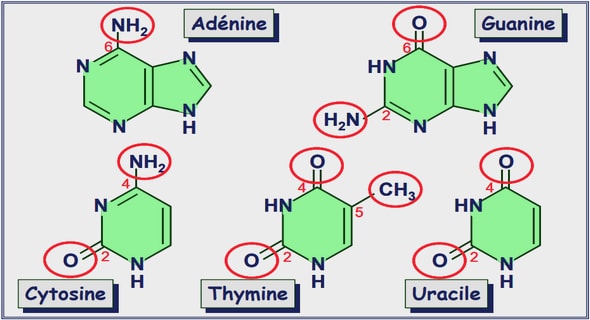
Synthèse d’oligodésoxynucléotides
La synthèse des acides désoxyribonucléiques est réalisée avec un synthétiseur Gene Assembler (Pharmacia). La séquence d’ADN est synthétisée de l’extrémité 3’ vers l’extrémité 5’ (Blackburn Gait, 1990), à partir des dérivés β-cyanoéthyle phosphoramidites des nucléosides. Le support solide utilisé est constitué de billes de silice sur lesquelles est greffé le premier nucléotide. La réaction de couplage est conduite en milieu anhydre dans l’acétonitrile, en présence de tétrazole comme activateur. Les synthèses sont réalisées à l’échelle de 2 µmol ou de 15 µmol. L’insertion d’une biotine dans la chaîne d’ADN est possible en utilisant une biotine amidite ou biodite (Pharmacia) comme base modifiée. La partie biotine de la molécule est séparée de la partie amidite par un lien d’une dizaine d’atomes à partir du groupement carboxyle de la chaîne latérale de la biotine. Nous avons généralement procédé à ce type d’insertion à l’extrémité 5’ de la séquence synthétisée. Les oligodésoxynucléotides synthétisés sont purifiés par passage sur colonne anionique QSepharose (Pharmacia), dans un tampon à 60 mM NaOH (pH 12). L’élution est réalisée à l’aide d’un gradient de NaCl optimisé selon la séquence de l’oligodésoxynucléotide à purifier, et suivie par mesure de l’absorbance à 280 nm (longueur d’onde choisie pour ne pas saturer le détecteur). Après neutralisation, l’échantillon est dessalé par dialyse. La concentration est déterminée à partir du coefficient d’extinction molaire établi en fonction des premiers voisins (Cantor et al., 1970) et de la mesure de l’absorbance à 265 nm. La biotine n’absorbant pas à 265 nm, elle n’affecte pas la mesure d’absorbance.
Marquage radioactif des oligodésoxynucléotides
Les oligodésoxynucléotides sont marqués radioactivement à l’aide de γATP33 P. La phosphorylation en 5’ est réalisée à l’aide de la T4-polynucléotide kinase (Roche). La réaction a Chapitre II. Matériels et Méthodes – 24 – lieu à 37°C pendant 30 minutes, selon les procédures communément utilisées (Sambrook et al., 1989). L’ADN phosphorylé est séparé de la polynucléotide kinase par précipitation dans l’éthanol. Le précipité est lavé avec de l’éthanol à 70% puis repris dans 30 à 50 µl de Tris (90 mM) citrate (90 mM) EDTA (2 mM) (= TCE 1x) à pH 7. L’efficacité de la réaction de phosphorylation est visualisée par migration sur couche mince d’un échantillon du milieu réactionnel prélevé avant l’incubation à 37°C, et d’un échantillon de même volume prélevé en fin d’incubation. Le γATP33 P non utilisé est visualisé au front de migration. La différence de signal relevée entre les deux échantillons permet d’établir le rendement de kination. Les séquences radioactivement marquées seront annotées d’un « * » par la suite.
Gel dénaturant (ADN marqué)
La qualité des échantillons d’acides nucléiques purifiés est contrôlée par migration électrophorétique dénaturante. L’échantillon préalablement marqué radioactivement est dénaturé à 90°C en présence d’urée (6M) pendant cinq minutes avant d’être déposé sur gel (acrylamide/bisacrylamide 12%, urée 6M). La migration se fait sur système d’électrophorèse non réfrigéré, à 30W (pour un gel de 20 cm x 20 cm x 0.8 mm). Après migration, les acides nucléiques sont fixés et l’urée éliminée dans un mélange acide acétique 10% – éthanol 20%. Le gel est séché sur papier Whatman. Les résultats sont obtenus après exposition nocturne et développement d’un film Kodak Biomax MR. Le rendement final de la synthèse d’ADN est généralement de 97 % pour une séquence de 20-25 bases, 75% si une biodite est insérée en fin de synthèse.