La neuromyélite optique de Devic
MECANISMES PHYSIOPATHOLOGIQUES
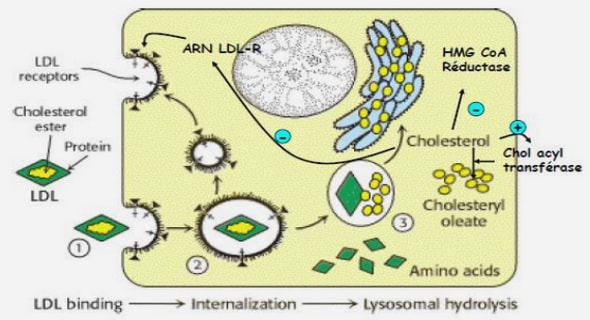
La découverte des anticorps anti-NMO et de leur cible, l’aquaporine 4 (AQP4) – canal hydrique le plus abondant du SNC-, fait de la NMO la première pathologie démyélinisante inflammatoire du SNC avec auto-anticorps identifié. Cependant, il est probable qu’elle ne soit pas seulement liée à la présence de ces anticorps. L’AQP4 est insérée dans la membrane cytoplasmique sous la forme d’un hétéro-tétramère, dont chaque monomère est composé de 323 acides aminés (isoforme M1) ou de 301 acides aminés (isoformes M23). Elle est localisée au niveau des podocytes des astrocytes des interfaces SNC-sang et SNC-LCR des espaces sous-arachnoïdiens sous-épendymaires et péri-capillaires (64). Elle est beaucoup plus fortement exprimée au niveau de la rétine, des nerfs et du tractus optiques. Il a été démontré une perte importante d’AQP4 au niveau des lésions médullaires de la NMO. L’interaction astrocyte /anti-NMO altère l’expression et la polarité de l’AQP4 et augmente la perméabilité de la barrière hémato-encéphalique. Les anti –NMO de type IgG peuvent servir d’activateur du complément lorsqu’ils se lient aux astrocytes. Ainsi, ils sont à l’origine de la libération d’anaphylatoxines de type C3a et C5a qui provoqueront un processus entrainant la mort des astrocytes. Cette mort semble être importante dans la physiopathologie de la NMO. Les patients atteints ont une diminution dans le LCR de la GFPA (Glial Fibrillaire Acide proteine) protéine marqueur des astrocytes, qui s’améliore sous traitement. Cependant, ces explications semblent insuffisantes pour expliquer la physiopathologie de la NMO. Chez ces patients tous les tissus contenant de l’AQP4 ne sont pas pathologiques (rein, épithélium intestinal). Par ailleurs, 40% des patients atteints de NMO n’ont pas d’anti-NMO.
DIAGNOSTIC
DIAGNOSTIC POSITIF
CLINIQUE
La clinique de la NMO a été précisée à travers plusieurs séries de la littérature. Le premier symptôme survient en général pendant la troisième décennie, avec des extrêmes entre 10 et 73 ans (9,36, 66, 68). La prépondérance féminine est massive, dépassant toujours 90 % des sujets. Aucune circonstance favorisante n’est étroitement associée même si des cas isolés débutent en post-partum (1, 5, 31, 55). Des épisodes pseudo-grippaux sont rapportés dans une proportion variable. Une atteinte d’emblée mixte optico-spinale survient dans un quart des cas en moyenne [9, 55, 66, 68], principalement dans les formes monophasiques [10]. Le délai nécessaire à réunir les critères optiques et médullaires est en moyenne de 2,3 ans, mais il est inférieur à 1 an pour 60 % des sujets (5). a- Syndrome oculaire Une névrite optique isolée est révélatrice chez 50 à 65 % des sujets, bilatérale dans 1/3 des cas (5, 36). L’atteinte est une NO rétrobulbaire, mais la neuropapillite est plus évocatrice. Les particularités cliniques sont la profondeur de la baisse d’acuité visuelle et la médiocrité de la récupération fonctionnelle puisque une cécité est atteinte après une moyenne de 1,6 événement par œil (5). Cependant, le premier épisode optique régresse totalement dans 60 % des cas (5). Des poussées purement douloureuses peuvent aussi se manifester au niveau de l’œil aveugle. À la phase d’état, une atrophie optique et un scotome absolu sont fréquemment observés.
Syndrome médullaire
Une localisation médullaire est le premier événement dans 20 à 50 % des cas de la littérature (8, 36, 66, 86). Le délai d’apparition de l’atteinte médullaire est en moyenne de 2 ans, et de 4,6 ans pour une myélite sévère (5). Des prodromes douloureux sont fréquemment rapportés à type de cervicalgies, dorsalgies ou céphalées mais n’ont pas fait l’objet d’étudeS spécifiqueS. La myélopathie s’installe sur un mode aigu atteignant l’acmé en quelques heures ou plus souvent quelques jours. Une installation en marche d’escalier étalée sur plusieurs semaines est parfois rapportée. La topographie est volontiers cervico-dorsale mais tous les niveaux peuvent être intéressés. Le tableau clinique varie avec le niveau et l’extension des lésions. La présentation typique est celle d’une myélite transverse symétrique. Le tableau peut être dramatique à type de tétraplégie flasque associée à une détresse respiratoire requérant une assistance ventilatoire. Les poussées médullaires peuvent également adopter une topographie plus limitée comme des syndromes de BrownSéquard, des monoparésies, voire des troubles sensitifs isolés. Lors des poussées cervicales, des signes supra-médullaires sont observés (down-beating nystagmus, hoquet, syndrome bulbaire ou cérébelleux) qui correspondent à une extension bulbo-protubérantielle des lésions qui ne récuse pas le diagnostic (59, 76). Un syndrome centromédullaire serait souvent observé (59, 82) sous la forme d’un syndrome extra-lemniscal suspendu, mieux individualisable à la phase d’état où il peut être le seul signe résiduel de même que des signes sympathiques par atteinte de la substance grise médullaire (82). À la phase d’état, la paralysie sur un mode plégique d’au moins un membre et son caractère flasque seraient fréquents. Des phénomènes paroxystiques douloureux et moteurs sont souvent rapportés à distance des poussées. 9 2)
PARACLINIQUE
Imagerie par Résonnance magnétique (IRM)
Elle s’intéresse à 2 structures du SYSTEME NERVEUX CENTRAL : la MOELLE EPINIERE et le cerveau. Les séquences standards (T1, T2 ou T2-FLAIR avec ou sans injection de Gadolinium) sont utilisées. Rarement d’autres séquences sont nécessaires (transfert d’aimantation, diffusion…). 1- IRM Médullaire C’est un examen clef pour le diagnostic. Un aspect de myélite transverse extensive est caractéristique. L’IRM en séquence T2 montre habituellement des hypersignaux (43). Au stade aigu, les lésions peuvent s’accompagner d’une prise de contraste (69%), souvent diffuse, avec un œdème (67%) et une nécrose sous la forme d’une cavitation (16%). L’extension verticale sur 3 segments vertébraux ou plus est le marqueur IRM le plus important de la maladie (43). La partie cervicale de la moelle est touchée dans prés de 2/3 des cas. Les poussées médullaires récidivent au même endroit ou à proximité dans 67% des cas. La partie centrale de la moelle épinière est la plus touchée. Le suivi en IRM permet de voir une diminution progressive de l’œdème et de l’hypersignal T2 avec parfois une fragmentation des anomalies de signal et parfois le développement d’une atrophie médullaire dans 13 à 22%. L’IRM de diffusion peut montrer une augmentation significative de la diffusivité moyenne des lésions médullaires par rapport à celles de la SEP. Le transfert de magnétisation peut être utile, comme indicateur non spécifique de l’intégrité tissulaire. Il y a une différence significative avec les sujets normaux témoins, alors qu’il n’y avait pas de différence avec les sujets atteints de la SEP avec une telle lésion (4,53). 10 Image1 : coupe médullaire coronale avec hypersignal T2 centromédullaire étendu. Image2 : coupes sagittales avec hypersignal T2 médullaire étendu Image3 : coupes médullaires coronale, et sagittales avec hypersignal médullaire. Image4 : coupe sagittale avec hypersignal T2 médullaire cervical Laplaud DA.
IRM cérébrale
Pendant longtemps, l’absence de lésions cérébrales était l’un des critères de la maladie. Des études récentes vont changer cette vision. 60 à 89% des cas auront des lésions cérébrales. Le commun des lésions est qu’elles sont aspécifiques et ne répondent pas aux critères de Barkhof ou de Paty. Des lésions spécifiques en T2 seront observées en periventriculaire, au niveau du tronc cérébral du cervelet dans 8% des cas. En séquence T1, on peut observer une prise de contraste décrite comme ressemblant à des nuages, sous la forme de petites zones adjacentes multiples, comme une accumulation de nuages. En séquences de diffusion, la fraction d’anisotropie est normale sauf dans la SB où elle parait légèrement diminuée.
Biologie du liquide céphalorachidien (LCR)
L’étude du LCR rapporte habituellement une pleiocytose avec une proteinorrachie. La pleiocytose est le plus souvent à prédominance lymphocytaire avec 23 +/-7,4 par mm3 et des polynucléaires aux environ de 9 +/- 14,7 par mm3 (18). Il existe une différence significative entre SEP et NMO concernant la présence de bandes oligoclonales en IgG (BOC). Ainsi, dans la NMO seuls 23 à 35% des patients présentent des BOC, alors que c’est le cas chez prés de 90% des SEP (18). Par ailleurs, ces BOC disparaissent souvent au cours de la maladie (43). Enfin, la réaction intrathécale poly spécifique lymphocytaire B (ou réaction MRZ pour oreillons, rubéole et zona) est habituelle dans la SEP mais absente dans la NMO (53). 12 c- Anticorps anti-NMO Découvert en 2004, Il s’agit d’auto- anticorps de type IgG, appelés anti-NMO, détectés à l’immunofluorescence indirecte (IFI), et permettant de différencier la NMO de la SEP. Alors que la spécificité du test est excellente (90 à 99,6%), sa sensibilité n’est pas aussi importante (50 à 73%). Leur présence semble de mauvais pronostic. Les patients séropositifs font plus de poussées, surtout médullaires et ont un score EDSS plus élevé que ceux séronégatifs (10, 42,57). Leur présence chez des patients présentant soit des névrites optiques à répétition, soit des myélites transverses à répétition est prédictive de l’évolution vers une NMO .
PREMIERE PARTIE : REVUE DE LA LITTERATURE |